
Filed Pursuant to Rule 424(b)(3)
Registration No. 333-260849
PROSPECTUS
PHARMACYTE BIOTECH, INC.
8,050,000 SHARES OF COMMON STOCK ISSUED
PURSUANT TO THE EXERCISE OF WARRANTS
This prospectus relates to the resale by the selling stockholders (“Selling Stockholders”) named herein, including their transferees, pledgees or donees, or their respective successors, of up to 8,050,000 shares (“Shares”) of PharmaCyte Biotech, Inc.’s (“Company”) common stock, $.0001 par value (“Common Stock”). For information about the Selling Stockholders, see “Selling Stockholders” on page 30. Our Common Stock offered by the Selling Stockholders will be issued pursuant to the exercise of warrants in accordance with their terms. The Selling Stockholders may sell shares of Common Stock from time to time in the principal market on which the Company’s Common Stock is quoted at the prevailing market price or in negotiated transactions. We are not selling any Common Stock under this prospectus and will not receive any of the proceeds from the sale of Common Stock by the Selling Stockholders. We will pay the expenses of registering these Shares, including legal and accounting fees. All selling and other expenses incurred by the Selling Stockholders will be borne by the Selling Stockholders. See “Plan of Distribution.”
The Shares offered by the Selling Stockholders are issuable pursuant to the exercise of 7,000,000 unregistered warrants (“Series A Warrants”) sold in a private placement and issued under that certain Securities Purchase Agreement dated as of August 19, 2021(“Purchase Agreement”) with each of the Selling Stockholders. Each Series A Warrant has an exercise price of $5.00 per share, is exercisable immediately, and will expire five years following the date of issuance. In addition, the offered Shares include 1,050,00 shares of Common Stock issuable through the exercise of 1,050,000 warrants (“Placement Agent Warrants,” and, together with the Series A Warrants, the “Warrants”) issued to H.C. Wainwright & Co., LLC (“Wainwright”) or its designees as the Company’s exclusive placement agent for the Purchase Agreement and a concurrent Registered Direct Offering (together, “Offerings”). The Placement Agent Warrants have an exercise price of $6.25 per share and will expire five years after the date of commencement of sales in the Offerings. See “Private Placement of Warrants” on page 28.
The timing and amount of any sale of Common Stock is within the sole discretion of each Selling Stockholder.
Our Common Stock is traded on the Nasdaq Stock Market (“Nasdaq”) under the symbol “PMCB”. On November 18, 2021, the closing sales price for our Common Stock on Nasdaq was $2.59 per share.
The purchase of the Common Stock offered through this prospectus involves a high degree of risk. You should consider carefully the risk factors beginning on page 14 of this prospectus before purchasing any of the securities offered by this prospectus.
We may amend or supplement this prospectus from time to time by filing amendments or supplements as required. You should read the entire prospectus and any amendments or supplements carefully before you make your investment decision.
Neither the Securities and Exchange Commission (“SEC”) nor any state securities commission has approved or disapproved of these securities or passed upon the adequacy or accuracy of this prospectus. Any representation to the contrary is a criminal offense.
The date of this prospectus is November 19, 2021.
TABLE OF CONTENTS
Page
You should rely only on the information provided in or incorporated by reference in this prospectus, in any prospectus supplement or in a related free writing prospectus, or documents to which we otherwise refer you. We have not authorized anyone else to provide you with different information.
We have not authorized any dealer, agent or other person to give any information or make any representation other than those contained or incorporated by reference in this prospectus and any accompanying prospectus supplement or any related free writing prospectus. You must not rely upon any information or representation not contained or incorporated by reference in this prospectus or an accompanying prospectus supplement or any related free writing prospectus. This prospectus and the accompanying prospectus supplement and any related free writing prospectus, if any, do not constitute an offer to sell or the solicitation of an offer to buy any securities other than the registered securities to which they relate, nor does this prospectus and the accompanying prospectus supplement and any related free writing prospectus, if any, constitute an offer to sell or the solicitation of an offer to buy securities in any jurisdiction to any person to whom it is unlawful to make such offer or solicitation in such jurisdiction. You should not assume that the information contained in this prospectus and the accompanying prospectus supplement and any related free writing prospectus, if any, is accurate on any date subsequent to the date set forth on the front of such document or that any information we have incorporated by reference is correct on any date subsequent to the date of the document incorporated by reference, even though this prospectus and any accompanying prospectus supplement and any related free writing prospectus is delivered or securities are sold on a later date.
We further note that the representations, warranties and covenants made by us in any agreement that is filed as an exhibit to any document that is incorporated by reference in this prospectus were made solely for the benefit of the parties to such agreement, including, in some cases, for the purpose of allocating risk among the parties to such agreements, and should not be deemed to be a representation, warranty or covenant to you. Moreover, such representations, warranties or covenants were accurate only as of the date when made. Accordingly, such representations, warranties and covenants should not be relied on as accurately representing the current state of our affairs.
Unless otherwise indicated, information contained or incorporated by reference in this prospectus concerning our industry, including our general expectations and market opportunity, is based on information from our own management estimates and research, as well as from industry and general publications and research, surveys and studies conducted by third parties. Management estimates are derived from publicly available information, our knowledge of our industry and assumptions based on such information and knowledge, which we believe to be reasonable. In addition, assumptions and estimates of our and our industry’s future performance are necessarily uncertain due to a variety of factors, including those described in “Risk Factors” beginning on page 14 of this prospectus. These and other factors could cause our future performance to differ materially from our assumptions and estimates.
In this prospectus the “Company,” “Registrant,” “PharmaCyte,” “we,” “us” and “our” refer to PharmaCyte Biotech, Inc., a Nevada corporation, and, where appropriate, to its subsidiaries.
We license the copyright for Cell-in-a-Box® in the United States from Austrianova Singapore Pte. Ltd. This prospectus contains references to our copyrights. Solely for convenience, copyrights and trade names referred to in this prospectus, including logos, artwork and other visual displays, may appear without the ® or TM symbols, but such references are not intended to indicate, in any way, that we will not assert, to the fullest extent under applicable law, our rights or the rights of the applicable licensor to these trademarks and trade names. We do not intend our use or display of other companies’ trade names or trademarks to imply a relationship with, or endorsement or sponsorship of us by, any other company.
| 1 |
This prospectus contains forward-looking statements. Forward-looking statements give our current expectations or forecasts of future events. You can identify these statements by the fact that they do not relate strictly to historical or current facts. Forward-looking statements involve risks and uncertainties and include statements regarding, among other things, our projected revenue growth and profitability, our growth strategies and opportunity, anticipated trends in our market and our anticipated needs for working capital. They are generally identifiable by use of the words “may,” “will,” “should,” “anticipate,” “estimate,” “plans,” “potential,” “projects,” “continuing,” “ongoing,” “expects,” “management believes,” “we believe,” “we intend” or the negative of these words or other variations on these words or comparable terminology. In particular, these include statements relating to future actions, prospective products, market acceptance, future performance or results of current and anticipated products, sales efforts, expenses, and the outcome of contingencies such as legal proceedings and financial results.
Examples of forward-looking statements in this prospectus include, but are not limited to, our expectations regarding our business strategy, business prospects, operating results, operating expenses, working capital, liquidity and capital expenditure requirements. Important assumptions relating to the forward-looking statements include, among others, assumptions regarding whether the United States Food and Drug Administration (“FDA”) approves our Investigational New Drug Application (“IND”) after we submit a response to the FDA’s clinical hold, so that we can commence our planned clinical trial involving locally advanced, inoperable, pancreatic cancer (“LAPC”); the success and timing of our preclinical studies and clinical trials; the potential that results of preclinical studies and clinical trials may indicate that any of our technologies and product candidates are unsafe or ineffective; our dependence on third parties in the conduct of our preclinical studies and clinical trials; the difficulties and expenses associated with obtaining and maintaining regulatory approval of our product candidates; the material adverse impact that the coronavirus pandemic may have on our business, including our planned clinical trial involving LAPC, which could materially affect our operations as well as the business or operations of third parties with whom we conduct business; and whether the FDA will approve our product candidates after our clinical trials are completed, assuming the FDA allows our clinical trials to proceed after submission and review of our response to the FDA’s clinical hold. Additional assumptions relate to the demand for our products, the cost, terms and availability of materials related to biopharma products, pricing levels, the timing and cost of capital expenditures, status of regulatory approvals, competitive conditions and general economic conditions. These statements are based on our management’s expectations, beliefs and assumptions concerning future events affecting us, which in turn are based on currently available information. These assumptions could prove inaccurate. Although we believe that the estimates and projections reflected in the forward-looking statements are reasonable, our expectations may prove to be incorrect.
Important factors that could cause actual results to differ materially from the results and events anticipated or implied by such forward-looking statements include, but are not limited to:
| · | our ability to conduct the preclinical studies and assays and provide the additional information requested by the FDA in order to lift the clinical hold on our IND for LAPC; |
| · | our ability to advance any product candidates into, and successfully complete, clinical studies and obtain regulatory approval for them; |
| · | the timing or likelihood of regulatory filings and approvals; |
| · | the commercialization, marketing and manufacturing of our product candidates, if approved; |
| · | the pricing and reimbursement of our product candidates, if approved; |
| · | the rate and degree of market acceptance and clinical utility of any products for which we receive marketing approval; |
| · | the implementation of our strategic plans for our business, product candidates and technology; |
| 2 |
| · | the scope of protection we have and are able to establish and maintain for intellectual property rights covering our product candidates and technology; |
| · | our expectations related to the use of proceeds from this offering and our existing cash resources, and estimates of our expenses, future revenues, capital requirements and our needs for additional financing; |
| · | our ability to maintain and establish collaborations; |
| · | our financial performance; |
| · | our ability to maintain compliance with Nasdaq listing standards; |
| · | developments relating to our competitors and our industry, including the impact of government regulation; |
| · | our ability to retain and attract senior management and consultants; and |
| · | other risks and uncertainties, including those listed under the caption “Risk Factors” in this prospectus or any other documents incorporated by reference in this prospectus. |
We operate in a very competitive and rapidly changing environment. New risks emerge from time to time. It is not possible for us to predict all of those risks, nor can we assess the impact of all of those risks on our business or the extent to which any factor may cause actual results to differ materially from those contained in any forward-looking statement. The forward-looking statements in this prospectus are based on assumptions management believes are reasonable. However, due to the uncertainties associated with forward-looking statements, you should not place undue reliance on any forward-looking statements. Further, forward-looking statements speak only as of the date they are made, and unless required by law, we expressly disclaim any obligation or undertaking to publicly update any of them in light of new information, future events, or otherwise.
Any of the assumptions underlying the forward-looking statements contained in this prospectus and the documents incorporated herein by reference could prove inaccurate and, therefore, we cannot assure you that the results contemplated in any of such forward-looking statements will be realized. Based on the significant uncertainties inherent in these forward-looking statements, the inclusion of any such statement should not be regarded as a representation or as a guarantee by us that our objectives or plans will be achieved, and we caution you against relying on any of the forward-looking statements contained herein.
This summary highlights certain information about us, this offering and information appearing elsewhere in this prospectus and in the documents we incorporate by reference in this prospectus. This summary is not complete and does not contain all of the information that you should consider before investing in our securities. After you carefully read this summary, to fully understand our Company and this offering and its consequences to you, you should read this entire prospectus and any related free writing prospectus authorized by us, including the information referred to under the heading “Risk Factors” in this prospectus beginning on page 14, and any related free writing prospectus, as well as the other documents that we incorporate by reference into this prospectus, including our financial statements and the notes to those financial statements, which are incorporated herein by reference from our Annual Report on Form 10-K for the year ended April 30, 2021, filed on August 10, 2021, and our Quarterly Report filed on Form 10-Q for the quarter ended July 31, 2021, filed on September 14, 2021. Please read “Where You Can Find More Information” on page 37 of this prospectus.
| 3 |
Overview
We are a biotechnology company focused on developing cellular therapies for cancer and diabetes based upon a proprietary cellulose-based live cell encapsulation technology known as “Cell-in-a-Box®.” The Cell-in-a-Box® technology is intended to be used as a platform upon which therapies for several types of cancer, including locally advanced, inoperable, pancreatic cancer (“LAPC”) will be developed. The current generation of our product candidate is referred to as “CypCaps™.” On October 1, 2020, we submitted an Investigational New Drug Application (“IND”) to the U.S. Food and Drug Administration (“FDA”) for a planned Phase 2b clinical trial in LAPC. On September 1, 2020, the Company received notice from the FDA that it had placed the IND on clinical hold. On October 30, 2020, the FDA sent a letter to us setting forth the reasons for the clinical hold and specific guidance on what we must do to have the clinical hold lifted. To lift the clinical hold, the FDA has informed us that we need to conduct additional preclinical studies and assays. The FDA also requested additional information regarding several topics, including DNA sequencing data, manufacturing information and product release specifications. We are in the process of conducting these studies and assays and gathering additional information to submit to the FDA. See “Our Investigational New Drug Application and the Clinical Hold” below.
The Cell-in-a-Box® encapsulation technology potentially enables genetically engineered live human cells to be used as a means to produce various biologically active molecules. The technology is intended to result in the formation of pinhead sized cellulose-based porous capsules in which genetically modified live human cells can be encapsulated and maintained. In a laboratory setting, this proprietary live cell encapsulation technology has been shown to create a micro-environment in which encapsulated cells survive and flourish. They are protected from environmental challenges, such as the sheer forces associated with bioreactors and passage through catheters and needles, which we believe enables greater growth and production. The capsules are largely composed of cellulose (cotton) and are bio inert.
We are developing therapies for pancreatic and other solid cancerous tumors by using genetically engineered live human cells that we believe are capable of converting a cancer prodrug into its cancer-killing form. We encapsulate those cells using the Cell-in-a-Box® technology and place those capsules in the body as close as possible to the tumor. In this way, we believe that when a cancer prodrug is administered to a patient with a particular type of cancer that may be affected by the prodrug, the killing of the patient’s cancerous tumor may be optimized.
In addition, we have been exploring ways to delay the production and accumulation of malignant ascites fluid that results from many types of abdominal cancerous tumors. Malignant ascites fluid is secreted by abdominal cancerous tumors into the abdomen after the tumors have reached a certain stage of growth. This fluid contains cancer cells that can seed and form new tumors throughout the abdomen. This fluid accumulates in the abdominal cavity, causing swelling of the abdomen, severe breathing difficulties and extreme pain.
We have also been developing a potential therapy for Type 1 diabetes and insulin-dependent Type 2 diabetes. Our product candidate for the treatment of diabetes consists of encapsulated genetically modified insulin-producing cells. The encapsulation will be done using the Cell-in-a-Box® technology. Implanting these cells in the body is designed to function as a bio-artificial pancreas for purposes of insulin production.
We have also been considering ways to exploit the benefits of the Cell-in-a-Box® technology to develop therapies for cancer that involve prodrugs based upon certain constituents of the Cannabis plant; these constituents are of the class of compounds known as “cannabinoids”.
Until: (i) the FDA allows us to commence a clinical trial in LAPC described in our IND for which the FDA has placed a clinical hold; and (ii) we validate our Cell-in-a-Box® encapsulation technology in our planned Phase 2b clinical trial in LAPC, we are not spending any further resources developing this cannabinoid program.
Cancer Therapy
Targeted Chemotherapy
Our live-cell encapsulation technology-based potential therapies consist of encapsulated genetically modified living cells, with the type of encapsulated cell dependent on the disease being treated. For our lead product candidate, a therapy for pancreatic cancer, we propose that approximately 15,000-20,000 genetically modified live cells that produce an enzyme (an isoform of cytochrome P450), which we believe will convert the chemotherapy prodrug ifosfamide into its cancer-killing form, will be encapsulated using the Cell-in-a-Box® technology. In the clinical trial, if the FDA allows us to proceed, approximately 300 of these capsules will be placed in the patients’ blood supply and guided into place using interventional radiography so that they finally reside as close to the tumor in the pancreas as possible. Low doses (one gram per square meter of body surface area of the patient) of the chemotherapy prodrug ifosfamide will then be given to the patient intravenously.
| 4 |
The prodrug ifosfamide is normally activated in the patient’s liver. By activating the prodrug near the tumor using the Cell-in-a-Box® capsules, we believe our cellular therapy will act as a type of “bio-artificial liver.” Using this type of “targeted chemotherapy,” we are seeking to create an environment that enables optimal concentrations of the “cancer-killing” form of ifosfamide at the site of the tumor. Because the cancer-killing form of ifosfamide has a short biological half-life, we believe that this approach will result in little to no collateral damage to other organs in the body. We also believe this treatment will significantly reduce tumor size with no treatment-related side effects.
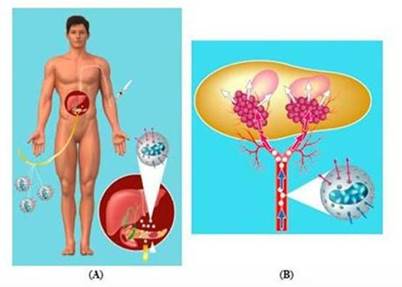
Figure 1: Proposed treatment for pancreatic cancer by targeted deployment and activation of chemotherapy using Cell-in-a-Box® encapsulated cells.
|
Note: Charts A and B are generalized graphic depictions of the principal hypothesized mechanisms of our proposed treatment for pancreatic cancer using our product candidate, the combination of Cell-in-a-Box® encapsulated cells plus low-doses of ifosfamide, under expected conditions. This combination therapy will be the subject of a clinical trial we plan to conduct, subject to FDA approval allowing us to move forward with our clinical trial. No regulatory authority has granted marketing approval for the Cell-in-a-Box® technology, the related encapsulated cells, or Cell-in-a-Box® and encapsulated cells plus low-dose ifosfamide combination. |
 |
|
Chart (A)
Cell-in-a-Box® capsules containing
live ifosfamide-activating cells (shown in white) will be implanted in the blood vessels leading to the tumor in the pancreas. Then low
dose ifosfamide will be given intravenously. |
Chart (B)
Chart B shows the human pancreas and generalized depictions of two pancreatic cancer tumors (shown in pink) as examples. In this chart, ifosfamide is converted to its cancer-killing form by the encapsulated live cells implanted near the tumors (shown in maroon).
Legend Blue Arrows: Ifosfamide enters capsules Red Arrows: Conversion to active form White Arrows: Activated ifosfamide targets tumors |
| 5 |
Figure 2: Hypothesized mechanism of action of treatment for pancreatic cancer by targeted deployment of the encapsulated live cells and activation of the chemotherapy prodrug drug ifosfamide. The immune system cells are too large to enter the capsule.
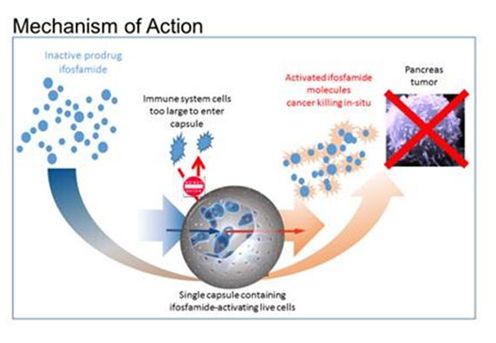
Pancreatic Cancer Therapy
We believe an unmet medical need exists for patients with LAPC whose pancreas tumor no longer responds after 4-6 months of treatment with either Abraxane® plus gemcitabine or the 4-drug combination known as FOLFIRINOX (folinic acid, fluorouracil, irinotecan and oxaliplatin). Both combinations are the current standards of care for pancreatic cancer. We believe that these refractory patients have no effective treatment alternative once their tumors no longer respond to these therapies. Two of the most commonly used treatments for these patients are 5-fluorouiracil (“5-FU”) or capecitabine (a prodrug of 5-FU) plus radiation (chemoradiation therapy). We believe that both treatments are only marginally effective in treating the tumor and both result in serious side effects. More recently, radiation treatment alone is being used at some cancer centers in the United States (“U.S.”).
Other treatments are being tried at various cancer centers in the U.S. in an attempt to address this lack of an effective treatment for many LAPC patients, but their success is far from certain. We are developing a therapy comprised of Cell-in-a-Box® encapsulated live cells implanted near the pancreas tumor followed by the infusion of low doses of the cancer prodrug ifosfamide. We believe that our therapy, if approved, can serve as a “consolidation therapy” that can be used with the current standards of care for LAPC and thus address this critical unmet medical need. Two previous human clinical trials of an encapsulated live cell and ifosfamide combination for LAPC were conducted in Germany by Bavarian Nordic during 1998 – 2000, and such trials were referenced in our IND for LAPC, submitted on October 1, 2020.
Subject to the FDA allowing us to move forward, we plan to commence a clinical trial involving patients with LAPC whose tumors have ceased to respond to either Abraxane® plus gemcitabine or FOLFIRINOX after 4-6 months of either therapy. The trial would initially take place in the U.S. with possible study sites in Europe at a later date.
Our Investigational New Drug Application and the Clinical Hold
On September 1, 2020, we submitted an IND to the FDA for a planned Phase 2b clinical trial in LAPC. Shortly thereafter, we received Information Requests from the FDA related to the IND. We timely responded to all Information Requests.
| 6 |
On October 1, 2020, we received notice that the FDA had placed our IND on clinical hold.
On October 30, 2020, the FDA sent a letter to us setting forth the reasons for the clinical hold and providing specific guidance on what we must do to have the clinical hold lifted.
In order to address the clinical hold, the FDA has requested that we:
| · | Provide additional sequencing data and genetic stability studies; |
| · | Conduct a stability study on the final formulated drug product candidate as well as the cells from our Master Cell Bank; |
| · | Evaluate the compatibility of the delivery devices (the prefilled syringe and the microcatheter used to implant the CypCaps™) with our drug product candidate; |
| · | Provide additional detailed description of the manufacturing process; |
| · | Provide additional product release specifications for our encapsulated cells; |
| · | Demonstrate comparability between the 1st and 2nd generation products and ensure adequate and consistent product performance and safety between the two generations of product; |
| · | Conduct a biocompatibility assessment using the final finished capsules after the entire drug product candidate manufacturing process (but without cells); |
| · | Address insufficiencies in Chemistry, Manufacturing and Controls information in the cross-referenced Drug Master File; |
| · | Conduct an additional nonclinical study in a large animal (such as a pig) to assess the safety, activity and distribution of the drug product candidate; and |
| · | Revise the Investigators Brochure to include any additional preclinical studies conducted in response to the clinical hold and remove any statements not supported by the data. |
The FDA also requested that we address the following issues as an amendment to the IND:
| · | Provide a Certificate of Analysis for pc3/2B1 plasmid that includes tests for assessing purity, safety, and potency; |
| · | Perform qualification studies for the drug substance filling step to ensure that the product candidate remains sterile and stable during the filling process; |
| · | Submit an updated batch analysis for the drug product candidate for the specific lot that will be used for manufacturing all future drug product candidate; |
| · | Provide additional details for the methodology for the Resorufin (CYP2B1) potency and the PrestoBlue cell metabolic assays; |
| · | Provide a few examples of common microcatheters that fit the specifications in our Angiography Procedure Manual; |
| · | Clarify the language in the Pharmacy Manual regarding proper use of the syringe fill with the drug product candidate; and |
| · | Provide a discussion with data for trial of the potential for cellular and humoral immune reactivity against the heterologous rat CYP2B1 protein and potential for induction of autoimmune-mediated toxicities in our study population in the LAPC. |
| 7 |
We have assembled a scientific and regulatory team of experts to address the FDA requests. That team is working to complete the items requested by the FDA. We are in varying stages of addressing the studies and acquiring the information requested by the FDA.
The following provides a summary of the activities in which we are engaged to have the clinical hold lifted:
| · | We have completed a 3, 6, 9, 12 and 18-month product stability study of our clinical trial product (CypCaps™), including container closure integrity testing for certain timepoints; the next time point in this ongoing study will be at 24 months of product stability. |
| · | We are involved in various additional studies required by the FDA. These include (i) a stability study on the cells from our Master Cell Bank (“MCB”) used to make the CypCaps™, which are already at the 3-year stability timepoint; (ii) further sequence analysis of the DNA encoding of the Cyp2B1 gene in the cells in the CypCaps™; and (iii) collated existing information on the reproducibility and quality of the filling of the MCB cells into vials ready for CypCaps™ manufacturing. |
| · | We are also involved in a (i) Subchronic and Chronic Toxicity study (ii) a Skin Sensitization study; (iii) an Acute Systematic Toxicity study; (iv) an Ames test (Genotoxicity Bacteria and Reverse Mutation tests); (v) an Intracutaneous test; (vi) a Complement Activation test; (vii) a Hemolysis test; (viii) an In Vitro Cytotoxicity test; and (ix) an In Vivo Micronucleus assay. Some of the data being generated by these studies will also be used to demonstrate comparability with the CypCaps™ that were used in the two earlier German clinical trials over twenty years ago conducted by Bavarian Nordic. |
| · | To enable the biocompatibility studies to be performed, we had Austrianova manufacture and deliver an additional 400 syringes of empty capsules. |
| · | We have commenced studies to show that CypCaps™ are not in any way adversely affected by the catheters used by interventional radiologists to deliver them, nor by the contract media used to visualize the blood vessels during implantation of the CypCaps™. |
| · | We have commenced studies to demonstrate how robust the CypCaps™ are during delivery and use as well as to document that the syringes used to deliver the CypCaps™ will allow delivery consistently, smoothly and safely. |
| · | With our support, Austrianova is providing additional detailed confidential information to the FDA on the manufacturing process, including information on the improvements made to the live cell encapsulated product since the last clinical trials with respect to reproducibility and safety of the CypCaps™. |
| · | We plan to update our IND submission documents to include: (i) more pre-clinical data as discussed above, (ii) some additional parameters for release of the CypCaps™, (iii) a recommendation of the catheters and contrast medium to be used to deliver the CypCaps™; and (iv) an extensive discussion of the potential for cellular and humoral immune reactivity against the heterologous rat CYP2B1 protein and potential for induction of autoimmune-mediated toxicities in our study population in the LAPC. |
| · | We have designed an abbreviated study in pigs to address biocompatibility and long-term implantation of the capsules. This animal study will complement the positive data already available from the previous human clinical trials conducted by Bavarian Nordic showing the safety of CypCaps™ implantation for up to two years in humans. |
| · | We have completed the complement activation study. The study results demonstrated that the capsule material we use does not activate a major line of the human body’s innate defense – the complement system. |
| 8 |
| · | Another positive result from a completed biocompatibility showed that the empty capsule material is “non-hemolytic.” |
| · | Another completed biocompatibility study showed that the empty capsule material is not “mutagenic.” |
Malignant Ascites Fluid Therapy
We have been exploring ways to delay the production and accumulation of malignant ascites fluid that results from many types of abdominal tumors. Malignant ascites fluid is secreted by an abdominal tumor into the abdomen after the tumor reaches a certain stage of growth. This fluid contains cancer cells that can seed and form new tumors throughout the abdomen. As this ascites fluid accumulates in the abdominal cavity, it can cause gross swelling of the abdomen, severe breathing difficulties and extreme pain.
Once an abdominal tumor reaches a certain stage of development, the tumor secretes malignant ascites fluid into the abdominal cavity. When that occurs, malignant ascites fluid must be removed by paracentesis on a periodic basis. This procedure is painful and costly. We know of no available therapy that prevents or delays the production and accumulation of malignant ascites fluid.
Preclinical studies were conducted by Translational Drug Development (“TD2”), an early-stage Clinical Research Organization (“CRO”) specializing in oncology, to examine whether the combination of Cell-in-a-Box® encapsulated cells plus low doses of ifosfamide can delay the production and accumulation of malignant ascites fluid. We believe the data from these studies support our plans to further explore whether the treatment might play a role in malignant ascites fluid production and accumulation. However, the conclusions were difficult to interpret with certainty. As a result, we plan to conduct another preclinical study in Germany to determine if our conclusions from the TD2 studies are valid. If this study is successful, and subject to discussions with the FDA, we plan to submit an IND to seek approval from the FDA to conduct a Phase 1 clinical trial in the U.S. to determine if our drug product candidate can delay the production and accumulation of malignant ascites fluid.
Diabetes Therapy
A Bio-Artificial Pancreas to Treat Diabetes
We are developing a therapy for Type 1 diabetes and insulin-dependent Type 2 diabetes based upon the encapsulation of a human liver cell line genetically engineered to produce, store and secrete insulin at levels in proportion to the levels of blood sugar in the human body. We are also considering an alternative route to bringing a biological treatment for diabetes into the clinic. We are exploring the possibility of encapsulating human insulin-producing cells and then transplanting them into a diabetic patient. Our plans are subject to discussions with the FDA.
The cell line we select will be encapsulated using the Cell-in-a-Box® encapsulation technology. If appropriate animal testing is completed successfully, and subject to discussions with the FDA, we intend to submit an IND to seek the FDA’s approval to transplant encapsulated insulin-producing cells into diabetic patients. The goal for these approaches is to develop a bio-artificial pancreas for purposes of insulin production for diabetics who are insulin-dependent.
Our diabetes program began with two of the most critical components of a biological diabetes therapy - a line of human cells which release insulin in response to the blood glucose level in their environment and a technology to protect the cells from an attack by the immune system once they are transplanted into a patient’s body to replace the patient’s own destroyed insulin-producing cells. This technology is the Cell-in-a-Box® encapsulation technology. The cells used are called Melligen cells. They are patent-protected and have been licensed to us by University of Technology Sydney (“UTS”).
Regulations for the use of living cells as a medical product require that the potential of the cells to grow and form a tumor in a patient be assessed. This so-called “tumorigenicity study” has been completed by the University of Veterinary Medicine Vienna (“VetMed”). Melligen cells showed very low tumorigenicity at a level we believe would expect to pass regulatory scrutiny, although this is subject to discussions with the FDA.
| 9 |
Putting Melligen cells and the Cell-in-a-Box® technology together, we conducted the first functional study in diabetic mice. The results did not meet our expectations. We discovered that, contrary to what we had expected and what we had read in published scientific papers on the Melligen cells published by UTS, the cells are not stable. With extensive testing and experiments, we discovered that the Melligen cells lose some of their specific beneficial properties over time.
We entered into a new research agreement with UTS to create an advanced version of the Melligen cells for the treatment of diabetes. Under the new research agreement, improvements will be made to the Melligen cells that we believe will increase their stability, increase their insulin production and increase the bioactivity of the produced insulin.
Prof. Ann Simpson, who created the Melligen cells, and her team of research scientists at UTS have been conducting this research project. The work is being funded by the Company and UTS. Our portion of the funding was previously paid to UTS. The research to date has not produced the results we had anticipated and is taking longer than we anticipated. It remains to be seen whether the Melligen cells are capable of producing the required insulin to be a viable cell line for the treatment of diabetes.
Cannabinoids to Treat Cancer
Numerous studies have demonstrated the therapeutic potential of certain cannabinoids (constituents of Cannabis) in patients with cancer. Two of the most widely studied cannabinoids in this regard are tetrahydrocannabinol (“THC”) and cannabidiol (“CBD”). Cannabinoids are potentially: (i) anti-proliferative (slow tumor growth); (ii) anti-metastatic (slow tumor spread); (iii) anti-angiogenic (slowing blood vessel development); and (iv) pro-apoptotic (initiate programmed cell death). In in vitro and in vivo models, the therapeutic potential of cannabinoids is broad. Results support the therapeutic potential in lung, brain, thyroid, lymphoma, liver, skin, pancreas, uterus breast and prostate cancers. In a review of 51 scientific studies, among other properties, it was observed that cannabinoids can regulate cellular signaling pathways critical for cell growth and survival. These properties indicate that cannabinoids could be useful in the treatment of cancer.
We have many competitors that are developing Cannabis-based treatments for cancer. Jazz Pharmaceuticals has acquired GW Pharmaceuticals, PLC who had an approved cannabinoid product for the treatment of multiple sclerosis spasticity and was developing a product portfolio to treat a variety of illnesses, including glioblastoma (brain cancer). Cannabis Science, Inc. has been developing topical cannabinoid treatments for basal and squamous cell skin cancers and Kaposi’s sarcoma, and is exploring pre-clinical development of cannabinoid-based anti-cancer drugs in a collaborative agreement with other entities. OWC Pharmaceutical Research Corp. is developing Cannabis-based products targeting a variety of indications and has a collaborative agreement with an academic medical center in Israel to study the effects of cannabinoids on multiple myeloma (a cancer of plasma cells). Cannabis Pharmaceuticals, Inc. is developing personalized anti-cancer and palliative Cannabis-based treatments aimed mainly at improving the cachexia, anorexia syndrome and quality-of-life issues that are often characteristic of patients with devastating diseases like cancer.
In contrast to the work being done by these companies, we plan to focus on developing specific therapies based on chosen molecules rather than using complex Cannabis extracts. We intend to use the Cell-in-a-Box® technology in combination with genetically modified cell lines designed to activate cannabinoid molecules for the treatment of diseases and their related symptoms. Our initial target will be glioblastoma, a very difficult-to treat form of brain cancer.
In May 2014, we entered into a research agreement with the University of Northern Colorado (“UNC”). The goal of the original research was to develop methods for the identification, separation and quantification of constituents of Cannabis, some of which are prodrugs, which could potentially be used in combination with the Cell-in-a-Box® technology to treat cancer.
In January 2017, we entered into a second research agreement with UNC. The goal of this research is to assess the synthesis of the patG gene and its incorporation into a vector, transfection of human embryonic kidney cells using this vector and assessment of cannabinoic acid decarboxylase activity.
| 10 |
During 2017, UNC identified an organism whose genome contains the genetic code for production of an enzyme capable of activating a cannabinoid prodrug into its active cancer-killing form. Our Cannabis program now has two primary areas of focus. The first is evaluating the therapeutic potential of cannabinoids, such as THC and CBD, particularly in our main “target” tumor – glioblastoma. UNC’s laboratory research has confirmed that a purified cannabinoid showed a potent dose-dependent decrease in cell viability for various cancers, suggesting that this cannabinoid exhibits significant anti-proliferative effects (stops the growth and multiplication of cancer cells). This activity has been demonstrated in brain (glioblastoma), pancreas, breast, lung, colon and melanoma cancer cells. The second area of focus is in finding an enzyme capable of converting an inactive, side-effect-free, cannabinoid prodrug into its active cancer-killing form.
Clinically, targeted cannabinoid-based chemotherapy would be accomplished by implanting the encapsulated bio-engineered cells near the site of a tumor, along with administration of a cannabinoid prodrug which would become activated at the site of the tumor by an enzyme produced by the encapsulated cells. We believe this could lead to better efficacy than existing therapies with minimal treatment related adverse events.
Until: (i) the FDA allows us to commence a clinical trial in LAPC described in our IND for which the FDA has placed a clinical hold; and (ii) we validate our Cell-in-a-Box® encapsulation technology in our planned Phase 2b clinical trial in LAPC, we are not spending any further resources developing this program.
Impact of the COVID-19 Pandemic on our Operations
The coronavirus SARS-Cov2 pandemic (“COVID-19”) is causing significant, industry-wide delays in clinical trials. Although we are not yet in a clinical trial, we have filed an IND with the FDA to commence a clinical trial in LAPC. While the IND has been placed on clinical hold by the FDA, we have assessed the impact of COVID-19 on our operations. Currently, many clinical trials are being delayed due to COVID-19. There are numerous reasons for these delays. For example, patients have shown a reluctance to enroll or continue in a clinical trial due to fear of exposure to COVID-19 when they are in a hospital or doctor’s office. There are local, regional and state-wide orders and regulations restricting usual normal activity by people. These discourage and interfere with patient visits to a doctor’s office if the visit is not COVID-19 related. Healthcare providers and health systems are shifting their resources away from clinical trials toward the care of COVID-19 patients. The FDA and other healthcare providers are making product candidates for the treatment of COVID-19 a priority over product candidates unrelated to COVID-19. As of the date of this prospectus, the COVID-19 pandemic has had an impact upon our operations, although we believe that impact is not material. The impact primarily relates to delays in tasks associated with the preparation of the Company’s responses to the clinical hold, including all requested preclinical studies. There may be further delays in generating responses to the requests from the FDA related to the clinical hold.
As a result of the COVID-19 pandemic, commencement of our planned clinical trial to treat LAPC may be delayed beyond the lifting of the clinical hold by the FDA should that occur. Also, enrollment may be difficult for the reasons discussed above. In addition, after enrollment in the trial, if patients contract COVID-19 during their participation in the trial or are subject to isolation or shelter in place restrictions, this may cause them to drop out of our clinical trial, miss scheduled therapy appointments or follow-up visits or otherwise fail to follow the clinical trial protocol. If patients are unable to follow the clinical trial protocol or if the trial results are otherwise affected by the consequences of the COVID-19 pandemic on patient participation or actions taken to mitigate COVID-19 spread, the integrity of data from the clinical trial may be compromised or not be accepted by the FDA. This could further adversely impact or delay our clinical development program if the FDA allows it to proceed.
It is highly speculative in projecting the effects of COVID-19 on our proposed clinical development program and the Company generally. The effects of COVID-19 quickly and dramatically change over time. Its evolution is difficult to predict, and no one is able to say with certainty when the pandemic will subside.
Recent Developments
Certificate of Amendment to Articles of Incorporation
On June 30, 2021, at our Annual Meeting of Stockholders, our stockholders approved a Certificate of Amendment to our Articles of Incorporation to increase the number of authorized shares of Common Stock from 2,500,000,000 shares to 50,000,000,000 shares. Upon effectiveness of the 1:1,500 reverse stock split on July 12, 2021, the number of authorized shares of our Common Stock was reduced proportionately to 33,333,334 shares by operation of Nevada law and the number of outstanding shares of our Common Stock was reduced to 1,591,420 shares. As of the date of this prospectus, and giving effect to the offerings discussed below, the Company had 20,715,804 shares of Common Stock outstanding.
| 11 |
Reverse Stock Split
On June 30, 2021, our Board of Directors (“Board”) approved a reverse stock split of 1:1,500 of our authorized and our issued and outstanding shares of Common Stock effective on July 12, 2021 pursuant to a Certificate of Change filed in Nevada. Except as otherwise indicated, all share and per share information in this prospectus gives effect to the reverse stock split of the Company’s outstanding Common Stock, which was effected at a ratio of 1-for-1,500 shares as of 12:01 a.m. Eastern Time on Monday, July 12, 2021.
Nasdaq Listing
Our Common Stock began trading on Nasdaq on August 10, 2021, under the symbol “PMCB.” Prior to that, our Common Stock was quoted on the OTCQB Market under the symbol “PMCB,” and following the reverse stock split of our Common Stock effective as of July 12, 2021, and until August 6, 2021, the OTCQB Market Symbol for our Common Stock had temporarily been PMCBD.
August 2021 Underwritten Offering
On August 9, 2021, we entered into an underwriting agreement with Wainwright as underwriter in connection with a public offering of an aggregate of (i) 2,630,385 shares of Common Stock and 899,027 pre-funded warrants (“August 9 Pre-funded Warrants”) to purchase Common Stock, and (ii) Common Stock warrants (the “August Common Warrants”) to purchase 3,529,412 shares of Common Stock. Each share of Common Stock (or pre-funded warrant in lieu thereof) was sold together with an August Common Warrant to purchase one share of Common Stock at an effective combined public offering price of $4.25 per share of Common Stock and accompanying August Common Warrant, less underwriting discounts and commissions. The August Common Warrants have an exercise price of $4.25 per share, are exercisable immediately, and will expire five years following the date of issuance. The August Pre-funded Warrants have an exercise price of $0.001 per share, are exercisable immediately, and do not have an expiration date. In addition, the Company granted Wainwright a 30-day option (“Option”) to purchase up to 529,411 shares and/or August Common Warrants at the public offering price, less the underwriting discounts and commissions. The offering of such securities pursuant to the underwriting agreement (“August 2021 Offering”) closed on August 12, 2021, and at closing, Wainwright partially exercised its Option for warrants to purchase an aggregate of up to 499,116 shares of Common Stock. At the closing, we received net proceeds from the offering of approximately $13.6 million, after deducting underwriting discounts and commissions and estimated offering expenses.
August 2021 Registered Direct Offering
Pursuant to the August 19, 2021 Purchase Agreement, the Company agreed to sell in a registered direct offering (“Registered Direct Offering”) 8,430,000 shares (“Shares”) of the Company’s Common Stock and pre-funded warrants (“August 19 Pre-Funded Warrants”) to purchase up to 5,570,000 shares of Common Stock. The Pre-Funded Warrants have an exercise price of $0.001 per share and are immediately exercisable and can be exercised at any time after their original issuance until such August 19 Pre-Funded Warrants are exercised in full. The Registered Direct Offering of the Shares and the August 19 Pre-Funded Warrants was made pursuant to the Company’s shelf registration statement on Form S-3 (File No. 333-255044), declared effective by the Securities and Exchange Commission on April 14, 2021, and a related registration statement (File No. 333-258921) filed on August 19, 2021 in accordance with Rule 462(b) under the Securities Act of 1933, as amended (“Securities Act”), and a prospectus supplement that the Company has filed with the Securities and Exchange Commission relating to such securities. At closing, we received net proceeds of approximately $64 million after deducting placement fees and estimated offering expenses.
August 2021 Warrant Exercises
As of August 18, 2021, all 899,027 of the August 9 Pre-funded Warrants have been exercised.
| 12 |
As of August 31, 2021, 2,522,387 of the August Common Warrants have been exercised, for aggregate gross proceeds to the Company of $10,720,145.
As of August 31, 2021, 4,620,000 of the August 19 Pre-funded Warrants have been exercised for aggregate gross proceeds to the Company of $4,620.
Summary of Risks Associated with Our Business
Our business is subject to numerous risks and uncertainties that you should consider before investing in our company. These risks are described more fully in the section titled “Risk Factors” in this prospectus. These risks include, but are not limited to, the following:
| · | We are a biotechnology company with limited resources, a limited operating history and have no products approved for clinical trials or commercial sale, which may make it difficult to evaluate our current business and predict our future success and viability. |
| · | As a result of the clinical hold that has been placed on our IND by the FDA, it has taken and may continue to take considerable time and expense to respond to the FDA and no assurance can be given that the FDA will remove the clinical hold in which case our business and prospects will likely suffer material adverse consequences. |
| · | The recent and ongoing COVID-19 pandemic has affected and could continue to affect our operations, as well as the business or operations of third parties with whom we conduct business. Our business could be adversely affected by the effects of other future health pandemics in regions where we or third parties on which we rely have significant business operations. |
| · | If we are unable to successfully raise additional capital, our future clinical trials and product development could be limited and our long-term viability may be threatened. |
| · | Due to the significant resources required for the development of our programs, and depending on our ability to access capital, we must prioritize development of certain product candidates. We may expend our resources on programs that do not yield a successful product candidate and fail to capitalize on product candidates or indications that may be more profitable or for which there is a greater likelihood of success. |
| · | We currently have no commercial revenue and may never become profitable. |
| · | If we are unable to obtain, or if there are delays in obtaining, required approval from the applicable regulatory agencies, we will not be able to commercialize our product candidates and our ability to generate revenue will be materially impaired. |
| · | If allowed to proceed with our clinical development program, we intend to conduct clinical trials for certain of our product candidates at sites outside of the U.S., and the U.S. regulatory agencies may not accept data from trials conducted in such locations. |
| · | Promising results in previous clinical trials of our encapsulated live cell and ifosfamide combination for LAPC may not be replicated in future clinical trials which could result in development delays or a failure to obtain marketing approval. |
| · | We may not be able to protect our intellectual property rights throughout the world. |
| 13 |
| · | We rely and expect to continue to rely heavily on third parties to conduct our preclinical studies and clinical trials, if we are allowed to proceed with our planned clinical trial, and those third parties may not perform satisfactorily, including failing to meet deadlines for the completion of such studies and trials. |
| · | There was no consistent active trading market for our Common Stock prior to August 10, 2021, and public trading of our Common Stock may continue to fluctuate substantially. |
| · | A large number of shares of Common Stock may be issued and subsequently sold upon the exercise of existing options and warrants. |
| · | As a result of our Nasdaq listing, we will incur materially increased costs and become subject to additional regulations and requirements. |
| · | We may not be able to meet the continued listing requirements for Nasdaq or another nationally recognized stock exchange, which could limit investors’ ability to make transactions in our securities and subject us to additional trading restrictions. |
| · | We are a “smaller reporting company” under the SEC’s disclosure rules and have elected to comply with the reduced disclosure requirements applicable to smaller reporting companies. |
| · | As a non-accelerated filer, we are not required to comply with the auditor attestation requirements of the Sarbanes-Oxley Act. |
| · | Following the reverse stock split, the resulting market price of our Common Stock may not attract new investors, including institutional investors, and may not satisfy the investing requirements of those investors. Consequently, the trading liquidity of our Common Stock may not improve. |
Our Corporate Information
We are a Nevada corporation incorporated in 1996. In 2013, we restructured our operations to focus on biotechnology. The restructuring resulted in the Company focusing all of its efforts upon the development of a novel, effective and safe way to treat cancer and diabetes. In January 2015, the Company changed its name from Nuvilex, Inc. to PharmaCyte Biotech, Inc. to reflect the nature of its current business.
Our corporate headquarters are located at 3960 Howard Hughes Parkway, Suite 500, Las Vegas, Nevada 89169, and our telephone number is (917) 595-2850. We maintain a website at www.pharmacyte.com, to which we regularly post copies of our press releases as well as additional information about us. Our filings with the SEC will be available free of charge through the website as soon as reasonably practicable after being electronically filed with or furnished to the SEC. Information contained in our website is not a part of, nor incorporated by reference into, this prospectus or our other filings with the SEC, and should not be relied upon.
To date, we have had a limited operating history with our current business model and have not produced any revenues.
Investing in our Common Stock involves a high degree of risk. Before investing in our Common Stock, you should carefully consider the risks described below, together with all of the other information contained in this prospectus and incorporated by reference herein, including from our Annual Report on Form 10-K for the fiscal year ended April 30, 2021, as well as any amendment or update to our risk factors reflected in subsequent filings with the SEC. Some of these factors relate principally to our business and the industry in which we operate. Other factors relate principally to your investment in our securities. The risks and uncertainties described therein and below are not the only risks facing us. Additional risks and uncertainties not presently known to us or that we currently deem immaterial may also materially and adversely affect our business and operations.
| 14 |
If any of the matters included in the following risks were to occur, our business, financial condition, results of operations, cash flows or prospects could be materially and adversely affected. In such case, you may lose all or part of your investment.
We are a biotechnology company with limited resources, a limited operating history and have no products approved for clinical trials or commercial sale, which may make it difficult to evaluate our current business and predict our future success and viability.
We are a biotechnology company focused on developing cellular therapies for cancer based upon a proprietary cellulose-based live cell encapsulation technology known as “Cell-in-a-Box®.” In recent years, we have devoted substantially all our resources to the development of our product candidate for LAPC. We have limited resources, a limited operating history, no products approved for clinical trials or commercial sale and therefore have not produced any revenues. We have generated significant operating losses since our inception. Our net losses for the years ended April 30, 2021 and 2020 were approximately $3.6 million and $3.8 million, respectively. As of July 31, 2021, we had an accumulated deficit of approximately $108.4 million. Substantially all our losses have resulted from expenses incurred relating to our research and development programs and from general and administrative expenses and operating losses associated with our business.
We expect to continue to incur significant expenses and operating losses for the foreseeable future. We anticipate these losses will increase as we continue our research and development of, and, if approved by the FDA, commence clinical trials for, our product candidates. In addition to budgeted expenses, we may encounter unforeseen expenses, difficulties, complications, delays and other unknown factors that may adversely affect our business.
We have no facilities to conduct fundamental research and we have performed our research and development activities by collaboration with contract service providers and contract manufacturers, and by designing and developing research programs in collaboration with university-based experts who work with us to evaluate mechanism(s) of disease for which we have designed and developed product candidates. We have not maintained a principal laboratory or primary research facility for the development of our product candidates.
Biotechnology product development is a highly uncertain undertaking and involves a substantial degree of risk. We have not commenced or completed clinical trials for any of our product candidates, obtained marketing approval for any product candidates, manufactured a commercial scale product, or arranged for a third party to do so on our behalf, or conducted sales and marketing activities necessary for successful product commercialization. Given the highly uncertain nature of biotechnology product development, we may never commence or complete clinical trials for any of our product candidates, obtain marketing approval for any product candidates, manufacture a commercial scale product or arrange for a third party to do so on our behalf, or conduct sales and marketing activities necessary for successful product commercialization.
Our limited operating history as a company makes any assessment of our future success and viability subject to significant uncertainty. We will encounter risks and difficulties frequently experienced by early-stage biotechnology companies in rapidly evolving fields, and we have not yet demonstrated an ability to successfully overcome such risks and difficulties. If we do not address these risks and difficulties successfully, our business, operating results and financial condition will suffer.
As a result of the clinical hold that has been placed on our IND by the FDA, it has taken and may continue to take considerable time and expense to respond to the FDA and no assurance can be given that the FDA will remove the clinical hold in which case our business and prospects will likely suffer material adverse consequences.
On October 1, 2020, we received notice from the FDA that it had placed our IND for a planned Phase 2b clinical trial in LAPC on clinical hold. As part of the clinical hold process, the FDA has asked for additional information, tasks to be performed by us and new preclinical studies and assays. It has taken and may continue to take a considerable period of time, the length of which is not certain at this time, for us to conduct such tasks and preclinical studies and to generate and prepare the requested information. It is possible that the service providers that we will utilize for such work may have considerable backlogs and/or are suffering from slowdowns as a result of COVID-19 and may not be able to perform such work for an extended period of time. Even if we are able to fully respond to the FDA’s requests, they may subsequently make additional requests that we would need to fulfill prior to the lifting of the clinical hold and we may never be able to begin our clinical trial in LAPC, obtain regulatory approval or successfully commercialize our product candidates. An inability to conduct our clinical trial in LAPC as a result of the clinical hold or otherwise, would likely force us to terminate our clinical development plans. It is possible that we will be unable to fully respond to the FDA in a satisfactory manner, and as a result the clinical hold may never be lifted. If the clinical hold is not lifted or if the lifting takes an extended period of time, our business and prospects will likely suffer material adverse consequences
| 15 |
The recent and ongoing COVID-19 pandemic could materially affect our operations, as well as the business or operations of third parties with whom we conduct business. Our business could be adversely affected by the effects of other future health pandemics in regions where we or third parties on which we rely have significant business operations.
Our business and its operations, including, but not limited to, our proposed clinical development program, supply chain operations, research and development activities and fundraising activities, has been and could continue to be adversely affected by the COVID-19 pandemic in areas where we have business operations, including the U.S., India, Europe, Singapore and Thailand. Also, this pandemic could cause significant disruption in the operations of third parties upon whom we rely on to conduct the Company’s business. In March 2020, the World Health Organization declared the COVID-19 outbreak a pandemic. Shortly thereafter, the U.S. government-imposed restrictions on travel between the U.S., Europe, and certain other countries. The President of the U.S. declared the COVID-19 pandemic a national emergency. Since March 2020, numerous state, regional and local jurisdictions, including the jurisdictions where our headquarters are located, as well as foreign jurisdictions, have imposed, and others in the future may impose, quarantines, shelter-in-place orders, executive, and similar government orders for their residents to control the spread of COVID-19. The COVID-19 pandemic has had an impact upon our operations.
The effects of the executive orders, the shelter-in-place orders and our work-from-home policies has and may continue to negatively impact productivity, disrupt our business, and delay our proposed clinical development program and timeline, the magnitude of which will depend, in part, on the length and severity of the restrictions and other limitations on our ability to conduct our business in the ordinary course. These and similar, and perhaps more severe, disruptions in our operations could negatively impact our business, operating results and financial condition.
Quarantines, shelter-in-place, executive, and similar government orders, or the perception that such orders, shutdowns or other restrictions on the conduct of business operations could occur, related to COVID-19, could impact personnel at our third-party manufacturing facilities in Thailand, or the availability or cost of materials we use or require to conduct our business, including product development, which would disrupt our supply chain. Some of our suppliers and vendors of certain materials used in our operations and research and development activities are located in areas that are subject to executive orders and shelter-in-place orders. While many of these materials may be obtained from more than one supplier, port closures and other restrictions resulting from the COVID-19 pandemic may disrupt our supply chain or limit our ability to obtain sufficient materials to operate our business. To date, we are aware of certain suppliers for our research and development activities that have experienced operational delays directly related to the COVID-19 pandemic.
Depending upon the length of the COVID-19 pandemic and whether the FDA lifts the clinical hold on our IND, we anticipate our planned clinical trial in LAPC may be affected by the COVID-19 pandemic. If COVID-19 continues to spread in the U.S. and elsewhere, we may experience additional disruptions that could adversely impact our business and proposed clinical trial, including: (i) delays or difficulties in enrolling patients in our Phase 2b clinical trial if the FDA allows us to go forward with such trial; (ii) delays or difficulties in clinical site activation, including difficulties in recruiting clinical site investigators and clinical site personnel; (iii) delays in clinical sites receiving the supplies and materials needed to conduct our clinical trial, including interruption in global shipping that may affect the transport of our clinical trial product; (iv) changes in local regulations as part of a response to the COVID-19 pandemic which may require us to change the ways in which our clinical trial is to be conducted, which may result in unexpected costs, or to discontinue the clinical trial altogether, if allowed to proceed; (v) diversion of healthcare resources away from the conduct of clinical trials, including the diversion of hospitals serving as our clinical trial sites and hospital staff supporting the conduct of our clinical trial; (vi) interruption of key clinical trial activities, such as clinical trial site monitoring, due to limitations on travel imposed or recommended by federal or state governments, employers and others, or interruption of clinical trial subject visits and study procedures, the occurrence of which could affect the integrity of clinical trial data; (vii) risk that participants enrolled in our proposed clinical trials will acquire COVID-19 while the clinical trial is ongoing, which could impact the results of the clinical trial, including by increasing the number of observed adverse events; (viii) delays in necessary interactions with local regulators, ethics committees, and other important agencies and contractors due to limitations in employee resources or forced furlough of government employees; (ix) limitations in employee resources that would otherwise be focused on the conduct of our clinical trial because of sickness of employees or their families or the desire of employees to avoid contact with large groups of people; (x) refusal of the FDA to accept data from clinical trials in affected geographies; and (xi) interruption or delays to our clinical trial activities.
| 16 |
The spread of COVID-19, which has caused a widespread impact throughout the world, may materially affect us economically. The potential economic impact brought about by the COVID-19 pandemic, and the duration of such impact, is difficult to assess or predict. The pandemic has resulted in significant disruption of global financial markets, which could reduce our ability to access capital and negatively affect our future liquidity. Also, a recession or market correction resulting from the spread of COVID-19 and related government orders and restrictions could materially affect our business and the value of our Common Stock. The COVID-19 pandemic continues to evolve. The ultimate impact of the COVID-19 pandemic and the mitigation efforts to address it is highly uncertain and subject to change. We do not yet know the full extent of potential delays or impacts on our business, our proposed clinical trial, healthcare systems or the global economy.
If we are unable to successfully raise additional capital, our future clinical trials and product development could be limited and our long-term viability may be threatened.
We have experienced negative operating cash flows since our inception and have funded our operations primarily through sales of our equity securities. We may need to seek additional funds in the future through equity or debt financings, or strategic alliances with third parties, either alone or in combination with equity financings to complete our product development initiatives. These financings could result in substantial dilution to the holders of our Common Stock, or require contractual or other restrictions on our operations or on alternatives that may be available to us. If we raise additional funds by issuing debt securities, these debt securities could impose significant restrictions on our operations. Any such required financing may not be available in amounts or on terms acceptable to us, and the failure to procure such required financing could have a material and adverse effect on our business, financial condition and results of operations, or threaten our ability to continue as a going concern.
Our operating and capital requirements during this fiscal year and thereafter will vary based on several factors, including whether the FDA allows us to commence our planned clinical trial for LAPC, how quickly enrollment of patients in our such trial can be commenced, the duration of the clinical trial and any change in the clinical development plans for our product candidates and the outcome, timing and cost of meeting regulatory requirements established by the FDA and the EMA or other comparable foreign regulatory authorities.
Our present and future capital requirements will be significant and will depend on many factors, including:
| · | whether the FDA lifts the clinical hold on our IND filing for LAPC; |
| · | the progress and results of our development efforts for our product candidates; |
| · | the costs, timing and outcome of regulatory review of our product candidates; |
| · | the costs and timing of preparing, filing and prosecuting patent applications, maintaining and enforcing our intellectual property rights and defending any intellectual property-related claims; |
| · | the effect of competing technological and market developments; |
| · | market acceptance of our product candidates; |
| 17 |
| · | the rate of progress in establishing coverage and reimbursement arrangements with domestic and international commercial third-party payors and government payors; |
| · | the extent to which we acquire or in-license other products and technologies; and |
| · | legal, accounting, insurance and other professional and business-related costs. |
We may not be able to acquire additional funds on acceptable terms, or at all. If we are unable to raise adequate funds, we may have to liquidate some or all of our assets, or delay or reduce the scope of or eliminate some or all of our development programs. Further, if we do not have, or are not able to obtain, sufficient funds, we may be required to delay development or commercialization of our product candidates. We also may have to reduce the resources devoted to our product candidates or cease operations. Any of these factors could harm our operating results.
Due to the significant resources required for the development of our programs, and depending on our ability to access capital, we must prioritize development of certain product candidates. We may expend our resources on programs that do not yield a successful product candidate and fail to capitalize on product candidates or indications that may be more profitable or for which there is a greater likelihood of success.
We seek to maintain a process of prioritization and resource allocation to maintain an optimal balance between aggressively advancing lead programs and ensuring replenishment of our portfolio. Until such time, if ever, as the FDA lifts its clinical hold on our IND related to our planned Phase 2b clinical trial in LAPC and our Cell-in-a-Box® encapsulation technology is validated in our planned Phase 2b clinical trial, we have halted spending on behalf of our development program with respect to cannabinoids.
Due to the significant resources required for the development of our programs, we must focus our programs on specific diseases and decide which product candidates to pursue and advance and the amount of resources to allocate to each. Our decisions concerning the allocation of research, development, collaboration, management and financial resources toward particular product candidates or therapeutic areas may not lead to the development of any viable commercial product and may divert resources away from better opportunities. Similarly, our potential decisions to delay, terminate or collaborate with third parties in respect of certain programs may subsequently also prove to be suboptimal and could cause us to miss valuable opportunities. We may fail to capitalize on viable commercial products or profitable market opportunities, be required to forego or delay pursuit of opportunities with other product candidates or other diseases that may later prove to have greater commercial potential than those we choose to pursue, or relinquish valuable rights to such product candidates through collaboration, licensing or other royalty arrangements in cases in which it would have been advantageous for us to invest additional resources to retain sole development and commercialization rights. If we make incorrect determinations regarding the viability or market potential of any or all of our programs or product candidates or misread trends in the biotechnology industry, our business, prospects, financial condition and results of operations could be materially adversely affected.
We currently have no commercial revenue and may never become profitable.
Even if we can successfully achieve regulatory approval for our product candidates, we do not know what the reimbursement status of our product candidates will be or when any of these products will generate revenue for us, if at all. We have not generated, and do not expect to generate, any product revenue for the foreseeable future. We expect to continue to incur significant operating losses for the foreseeable future due to the cost of our research and development, preclinical studies and clinical trials and the regulatory approval process for our product candidates. The amount of future losses is uncertain and will depend, in part, on the rate of growth of our expenses.
Our ability to generate revenue from our product candidates also depends on numerous additional factors, including our ability to:
| · | successfully complete development activities, including the remaining preclinical studies and planned clinical trials for our product candidates; |
| 18 |
| · | complete and submit NDAs or BLAs to the FDA and MAAs to the EMA, and obtain regulatory approval for indications for which there is a commercial market; |
| · | complete and submit applications to, and obtain regulatory approval from, other foreign regulatory authorities; |
| · | manufacture any approved products in commercial quantities and on commercially reasonable terms; |
| · | develop a commercial organization, or find suitable partners, to market, sell and distribute approved products in the markets in which we have retained commercialization rights; |
| · | achieve acceptance among patients, clinicians and advocacy groups for any products we develop; |
| · | obtain coverage and adequate reimbursement from third parties, including government payors; and |
| · | set a commercially viable price for any products for which we may receive approval. |
We are unable to predict the timing or amount of increased expenses, or when or if we will be able to achieve or maintain profitability. Even if we can complete the processes described above, we anticipate incurring significant costs associated with commercializing our product candidates.
We face substantial competition, which may result in others discovering, developing or commercializing competing products before or more successfully than we do.
The development and commercialization of new drug products is highly competitive. We face competition with respect to our current product candidates. We will face competition with respect to any product candidates that we may seek to develop or commercialize in the future. Such competition may arise from major pharmaceutical companies, specialty pharmaceutical companies and biotechnology companies worldwide. There are several large pharmaceutical and biotechnology companies that currently market products or are pursuing the development of products for the treatment of the disease indications for which we are developing our product candidates. Some of these competitive products and therapies are based on scientific approaches that are entirely different from our approach. Potential competitors also include academic institutions, government agencies and other public and private research organizations that conduct research, seek patent protection and establish collaborative arrangements for research, development, manufacturing and commercialization.
Specifically, there are numerous companies developing or marketing therapies for cancer and diabetes, including many major pharmaceutical and biotechnology companies. Our commercial opportunity could be reduced or eliminated if our competitors develop and commercialize products that are safer, more effective, have fewer or less severe side effects, are more convenient or are less expensive than any products that we may develop. Our competitors also may obtain regulatory approval for their products more rapidly than we may obtain approval for ours, which could result in our competitors establishing a strong market position before we can enter the market.
Many of the companies against which we are competing or against which we may compete in the future have significantly greater financial resources and expertise in research and development, manufacturing, preclinical testing, conducting clinical trials, obtaining regulatory approvals and marketing approved products than we do. Mergers and acquisitions in the pharmaceutical and biotechnology sectors may result in even more resources being concentrated among a smaller number of our competitors. Smaller and other early-stage companies may also prove to be significant competitors, particularly through collaborative arrangements with large and established companies. These third parties compete with us in recruiting and retaining qualified scientific and management personnel, establishing clinical trial sites and patient registration for clinical trials, as well as in acquiring technologies complementary to, or necessary for, our programs.
| 19 |
Additional Risks Related to Regulatory Matters
If we are unable to obtain, or if there are delays in obtaining, required approval from the applicable regulatory agencies, we will not be able to commercialize our product candidates and our ability to generate revenue will be materially impaired.
Our product candidates must obtain marketing approval from the FDA for commercialization in the U.S. and from foreign regulatory agencies for commercialization in countries outside the U.S. The process of obtaining marketing approvals in the countries in which we intend to sell and distribute our product candidates is expensive and can take many years, if approval is obtained at all. This process can vary substantially based upon a variety of factors, including the type, complexity and novelty of the product candidates involved. Failure to obtain marketing approval for a product candidate will prevent us from commercializing that product candidate. To date, we have not received approval to market any of our product candidates from regulatory agencies in any jurisdiction. We have no experience in filing and supporting the applications necessary to gain marketing approvals and expect to rely on third-party contract research organizations to assist us in this process. Securing marketing approval requires the submission of extensive preclinical and clinical data and supporting information to the regulatory agencies for each product candidate to establish the product candidate’s safety and efficacy. Securing marketing approval also requires the submission of information about the product manufacturing process to, and inspection of manufacturing facilities by, the regulatory agencies.
Our product candidates may not be effective, may be only moderately effective or may prove to have undesirable or unintended side effects, toxicities or other characteristics that may preclude our obtaining marketing approval or prevent or limit commercial use. Regulatory agencies have substantial discretion in the approval process and may refuse to accept any application or may decide that our data are insufficient for approval and require additional preclinical, clinical or other studies. In addition, varying interpretations of the data obtained from preclinical and clinical testing could delay, limit or prevent marketing approval of a product candidate. Changes in marketing approval policies during the development period, changes in or the enactment of additional statutes or regulations, or changes in regulatory review for each submitted product application, may also cause delays in or prevent the approval of an application. New cancer drugs frequently are indicated only for patient populations that have not responded to an existing therapy or have relapsed after such therapies. If we experience delays in obtaining approval or if we fail to obtain approval of our product candidates, the commercial prospects for our product candidates may be harmed and our ability to generate revenues will be materially impaired.
If allowed to proceed with our clinical development programs, we intend to conduct clinical trials for certain of our product candidates at sites outside of the U.S., and the U.S. regulatory agencies may not accept data from trials conducted in such locations.
Although the FDA may accept data from clinical trials conducted outside the U.S., acceptance of this data is subject to certain conditions imposed by the regulatory agencies outside of the U.S. For example, the clinical trial must be well designed and conducted and performed by qualified investigators in accordance with ethical principles. The trial population must also adequately represent the population in the country in which the clinical trial is being conducted. The data must be applicable to the U.S. population and medical practice in the U.S. in ways that the FDA deems clinically meaningful. Generally, the patient population for any clinical trial conducted outside of the U.S. must be representative of the population for whom we intend to seek approval in the U.S.
In addition, while these clinical trials are subject to the applicable local laws, the FDA acceptance of the data will be dependent upon its determination that the trials also complied with all applicable U.S. laws and regulations. There can be no assurance that the FDA will accept data from trials conducted outside of the U.S. If the FDA does not accept the data from any of our clinical trials that we determine to conduct outside the U.S., it would likely result in the need for additional trials that would be costly and time-consuming and delay or permanently halt the development of our product candidate.
In addition, the conduct of clinical trials outside the U.S. could have a significant impact on us. Risks inherent in conducting international clinical trials include:
| · | Foreign regulatory requirements that could restrict or limit our ability to conduct our clinical trials; |
| · | Administrative burdens of conducting clinical trials under multiple foreign regulatory schemes; |
| 20 |
| · | Foreign exchange fluctuations; and |
| · | Diminished protection of intellectual property in some countries |
Promising results in previous clinical trials of our encapsulated live cell and ifosfamide combination for LAPC may not be replicated in future clinical trials which could result in development delays or a failure to obtain marketing approval.
Positive results in the previous Phase 1/2 and Phase 2 clinical trials of the encapsulated live cell and ifosfamide combination product may not be predictive of similar results in future clinical trials such as our planned Phase 2b clinical trial in LAPC for which the FDA has placed a clinical hold. The previous Phase 1/2 and Phase 2 clinical trials were done over twenty years ago in Germany by Bavarian Nordic and had a relatively limited number of patients in each trial. These trials resulted in outcomes that were not statistically significant and may not be representative of future results. In addition, interim results obtained after a clinical trial has commenced do not necessarily predict results in future clinical trials. Numerous companies in the pharmaceutical and biotechnology industries have suffered significant setbacks in late-stage clinical trials even after achieving promising results in early-stage clinical development. Our clinical trials, if allowed to proceed, may produce negative or inconclusive results and we may decide, or regulatory agencies may require us, to conduct additional clinical trials. Moreover, clinical data are often susceptible to varying interpretations and analyses, and many companies that believed their product candidates performed satisfactorily in preclinical studies and clinical trials have nonetheless failed to obtain the approval for their products by the regulatory agencies.
Risks related to our Intellectual Property
We may not be able to protect our intellectual property rights throughout the world.
Filing, prosecuting and defending patents or establishing other intellectual property rights to our product candidates in all countries throughout the world would be prohibitively expensive, and our intellectual property rights in some countries outside the United States can be less extensive than those in the United States or non-existent. For example, the Melligen cells are protected by patents only in the U.S. and Europe and we are only pursuing patent protection for our pancreatic cancer product candidate in the U.S., Australia and Canada.
Many companies have encountered significant problems in protecting and defending intellectual property rights in foreign jurisdictions. The legal systems of some countries do not favor the enforcement of patents and other intellectual property protection, which could make it difficult for us to stop the infringement of our patents or misappropriation of our intellectual property rights generally. Proceedings to enforce our patent and other intellectual property rights in foreign jurisdictions could result in substantial costs and divert our efforts and attention from other aspects of our business, could put our patents or intellectual property rights at risk of being invalidated or interpreted narrowly and our patent applications at risk of not issuing and could provoke third parties to assert claims against us. We may not prevail in any lawsuits that we initiate, and the damages or other remedies awarded, if any, may not be commercially meaningful
Many countries, including European Union countries, India, Japan and China, have compulsory licensing laws under which a patent owner may be compelled under specified circumstances to grant licenses to third parties. In those countries, we may have limited remedies if patents are infringed or if we are compelled to grant a license to a third party, which could materially diminish the value of those patents. This could limit our ability to pursue strategic alternatives, including identifying and consummating transactions with potential third-party partners, to further develop, obtain marketing approval for and/or commercialize our product candidates, and consequently our potential revenue opportunities.
Our intellectual property and data and market exclusivity may not be sufficient to block others from commercializing identical or competing products.
Our success depends in large part on our ability to obtain and maintain both intellectual property rights and data and market exclusivity for our product candidates in order to block others from commercializing identical or competing products. Establishing intellectual property rights includes filing, prosecuting, maintaining, and enforcing patents that cover our product candidates and variations of our product candidates and protecting our trade secrets and other proprietary information related to our product candidates from unauthorized use.
| 21 |
The foundational patents relating to the Cell-in-the-Box® technology that were formerly licensed from Bavarian Nordic/GSF covering capsules encapsulating cells expressing cytochrome P450 and treatment methods using the same expired on March 27, 2017. Currently, we do not have any issued patents in any countries covering our product candidate for the treatment of pancreatic cancer. We exclusively license from UTS patented Melligen cells, which cover our product candidate for the treatment of diabetes, which are issued in the U.S. and Europe and expire in August 2028. Currently, we do not have any issued patents or pending applications covering our product candidate for the treatment of cancer using cannabinoids or our product candidate for the treatment of malignant ascites fluid therapy. We may not be able to obtain protection for our product candidates or variations of our product candidates. Even if our owned and licensed patent applications issue as patents, they may not issue in a form that will provide us with any meaningful protection, prevent competitors from competing with us or otherwise provide us with any competitive advantage or our patents may expire before or shortly after our product candidate is approved. Our competitors may be able to circumvent our owned or licensed patents by developing similar or alternative technologies or products in a non-infringing manner.
Confidential know-how and trade secrets are only protectable to the extent a third party utilizes the confidential know-how or trade secret in an unauthorized manner; however, if a third party is able to independently duplicate the technology, such as through reverse engineering, without access to or use of our confidential know-how or trade secret, we would have no recourse.
In addition, data exclusivity that is provided through the BPCIA in the U.S. and equivalents in foreign countries is limited in both time and scope. The BPCIA bars the FDA from approving biosimilar applications for 12 years after an innovator biological product receives initial marketing approval, however it does not bar the FDA from approving an identical or similar product that is the subject of its own BLA. Finally, upon the approval of the first BLA for a biologic designated as an Orphan Drug for a specified indication, the sponsor of that BLA is entitled to 7 years of exclusive marketing rights in the U.S. for biologic for the particular indication unless the sponsor cannot assure the availability of sufficient quantities to meet the needs of persons with the disease. In Europe, this exclusivity is 10 years. However, Orphan Drug status for an approved indication does not prevent another company from seeking approval of a biologic that has other labeled indications that are not under orphan or other exclusivities. In addition, in the U.S., the FDA is not prevented from approving another biologic for the same labeled Orphan indication if the company can demonstrate that the other biologic is clinically superior to first approved product.
Even if we are able to obtain patents and maintain confidential information and trade secrets and obtain data and market exclusivity for our product candidates, our competitors may be able to develop and obtain approval of identical or competing products.
If we do not obtain patent and/or data exclusivity for our product candidates, our business may be materially harmed.
Our commercial success will largely depend on our ability to obtain and maintain patent and other intellectual property protection and/or data exclusivity under the BPCIA in the U.S. and other countries with respect to our proprietary technology, product candidates and our target indications. If we are unable to obtain patents covering our product candidates or obtain data and/or marketing exclusivity for our product candidates, our competitors may be able to take advantage of our investment in development and clinical trials by referencing our clinical and preclinical data to obtain approval of competing products, such as a biosimilar, earlier than might otherwise be the case.
If we are unable to protect the confidentiality of our trade secrets, our business and competitive position would be harmed.
In addition to seeking patents for some of our technology and product candidates, we also rely on trade secrets, including unpatented know-how, technology and other proprietary information, to maintain our competitive position. We seek to protect these trade secrets, in part, by entering non-disclosure and confidentiality agreements with parties who have access to them, such as our employees, corporate collaborators, outside scientific collaborators, contract manufacturers, consultants, advisors and other third parties. We seek to protect our confidential proprietary information, in part, by entering confidentiality and invention or patent assignment agreements with our employees and consultants; however, we cannot be certain that such agreements have been entered with all relevant parties.
| 22 |
Moreover, to the extent we enter such agreements, any of these parties may breach the agreements and disclose our proprietary information, including our trade secrets to unaffiliated third parties. We may not be able to obtain adequate remedies for such breaches. Enforcing a claim that a party illegally disclosed or misappropriated a trade secret is difficult, expensive and time-consuming and the outcome is unpredictable. In addition, some courts inside and outside the U.S. are less willing or unwilling to protect trade secrets. If any of our trade secrets were to be lawfully obtained or independently developed by a competitor, we would have no right to prevent them, or those to whom they communicate them, from using that technology or information to compete with us. If any of our trade secrets were to be disclosed to or independently developed by a competitor, our competitive position would be harmed.
The majority of the technology that we license and use for our product candidates is not protected by patents, but rather is based upon confidential know-how and trade secrets. Confidential know-how and trade secrets are only protectable to the extent a third party utilizes the confidential know-how or trade secret in an unauthorized manner; however, if a third party is able to independently duplicate the technology, such as through reverse engineering, without access to or use of our confidential know-how or trade secret, we would have no recourse.
Risks Related to Our Dependence on Third Parties
We rely heavily on third parties to conduct our preclinical studies and plan to rely on third parties to conduct our clinical trials, assuming they are allowed to proceed, and those third parties may not perform satisfactorily, including failing to meet deadlines for the completion of such studies and trials.
We currently rely heavily on third parties to conduct our preclinical studies and plan to rely on third parties to conduct our clinical trials, assuming they are allowed to proceed, including Austrianova in which we own an equity interest. We expect to continue to rely heavily on third parties, such as a CRO, a clinical data management organization, a medical institution, a clinical investigator and others to plan for and conduct our clinical trials. Our agreements with these third parties generally allow the third party to terminate our agreement with them at any time. If we are required to enter alternative arrangements because of any such termination, the introduction of our product candidates to market could be delayed.
Our reliance on these third parties for research and development (“R&D”) activities will reduce our control over these activities but will not relieve us of our responsibilities. For example, we design our clinical trials and will remain responsible for ensuring that each is conducted in accordance with the general investigational plan and protocol for the trial. Moreover, regulatory agencies require us to comply with current good manufacturing practices (“cGMP”) for conducting, recording and reporting the results of clinical trials to assure that data and reported results are credible and accurate and that the rights, integrity and confidentiality of trial participants are protected. Our reliance on third parties that we do not control does not relieve us of these responsibilities and requirements. We also are required to register ongoing clinical trials and post the results of completed clinical trials on a government-sponsored database of regulatory agencies within specified timeframes. Failure to do so can result in fines, adverse publicity and civil and criminal sanctions.
Furthermore, these third parties may also have relationships with other entities, some of which may be our competitors. If these third parties do not successfully carry out their contractual duties, meet expected deadlines or conduct our clinical trials in accordance with the requirements of a regulatory agency or our protocols, we will not be able to obtain, or may be delayed in obtaining, marketing approvals for our product candidates and will not be able to, or may be delayed in our efforts to, successfully commercialize our product candidates.
| 23 |
We expect to rely on third parties to store and distribute our product candidates for our clinical trials, if they are allowed to proceed. Any performance failure on the part of such third parties could delay clinical development or marketing approval of our product candidates or commercialization of our products, if approved, producing additional losses and depriving us of potential product candidate revenue. Our existing collaboration with universities and institutions is important to our business. If we are unable to maintain these collaborations, or if these collaborations are not successful, our business could be adversely affected.
We rely on numerous consultants for a substantial portion of our R&D related to our product candidates. If there are delays or failures to perform their obligations, our product candidates would be adversely affected. If our collaboration with these consultants is unsuccessful or is terminated, we would need to identify new research and collaboration partners for our preclinical and clinical development. If we are unsuccessful or significantly delayed in identifying new collaboration and research partners, or unable to reach an agreement with such a partner on commercially reasonable terms, development of our product candidates will suffer, and our business would be materially harmed.
Furthermore, if any of these consultants change their strategic focus, or if external factors cause any one of them to divert resources from our collaboration, or if any one of them independently develops products that compete directly or indirectly with our product candidates using resources or information it acquires from our collaboration, our business and results of operations could suffer.
We rely on Prof. Günzburg, Dr. Salmons and Dr. Löhr for the development of our product candidates. If they decide to terminate their relationship with us, we may not be successful in the development of our product candidates.
We rely on Prof. Walter H. Günzburg and Dr. Brian Salmons, officers of Austrianova, and Dr. Matthias Löhr, currently with the Karolinska Institute in Stockholm, Sweden, for the development of our product candidates. If they decide to terminate their relationship with us, we may not be successful in the development of our product candidates.
Prof. Günzburg, Dr. Salmons and Dr. Löhr are involved in almost all our scientific endeavors underway and being planned by us. These endeavors include preclinical and clinical studies involving our cancer therapy for LAPC to be conducted in the U.S., if allowed to proceed, and elsewhere on our behalf. They also provide professional consulting services to us through the respective consulting agreements we have entered with the consulting companies through which they provide services. The consulting agreements may be terminated for any reason at any time upon one party giving the other a written notice prior to the effective date of the termination. If that occurs, we may not be successful in the development of our product candidates which could have a material adverse effect on us.
Risks Related to this Offering
Substantial future sales or other issuances of our Common Stock could depress the market for our Common Stock.
Sales of a substantial number of shares of our Common Stock, or the perception by the market that those sales could occur, could cause the market price of our Common Stock to decline or could make it more difficult for us to raise funds through the sale of equity in the future.
In connection with our August 2021 Offering, our directors and executive officers entered into lock-up agreements for a period of 90 days following such offering. Our directors and executive officers may be released from such lock-up agreements prior to the expiration of the lock-up period at the sole discretion of Wainwright. Upon expiration or earlier release of the lock-up, our directors and executive officers may sell shares into the market, which could adversely affect the market price of shares of our Common Stock.
Future issuances of our Common Stock or our other equity securities could further depress the market for our Common Stock. We expect to continue costs associated with our R&D programs, such as preclinical studies, clinical trials, and the regulatory approval process for therapeutic candidates, and general and administrative costs associated with our operations, and to satisfy our funding requirements, we may need to sell additional equity securities. The sale or the proposed sale of substantial amounts of our Common Stock or our other equity securities may adversely affect the market price of our Common Stock and our stock price may decline substantially. Our stockholders may experience substantial dilution and a reduction in the price that they are able to obtain upon sale of their shares. New equity securities issued may have greater rights, preferences or privileges than our existing Common Stock.
| 24 |
There was no consistent active trading market for our Common Stock prior to August 10, 2021, and public trading of our Common Stock may continue to fluctuate substantially.
Our Common Stock only began trading on Nasdaq on August 10, 2021. There was no consistent active trading market for our Common Stock prior to August 10, 2021 and there is no assurance that the trading market for our Common Stock will become more active or liquid. Furthermore, there can be no assurance our market makers will continue to make a market in our Common Stock. As a result, no assurances can be given that you will be able to readily sell your Common Stock at a price equal to or above the price you paid. We cannot provide any assurance that an active and liquid trading market in our securities will develop or, if developed, that such market will continue.
Moreover, the trading price of our Common Stock has fluctuated substantially over the past few years, and there remains a significant risk that our Common Stock price may continue to fluctuate substantially in the future in response to various factors, including any material developments in the FDA approval process for our proposed Phase 2b clinical trial, material variations in our periodic operating results, departures or additions of management or other key personnel, announcements of acquisitions, mergers, share consolidations, or new technology or patents, new product developments, significant litigation matters, gain or loss of significant licensees, significant capital transactions, substantial sales of our Common Stock in our trading market, and general and specific market and economic conditions.
We may not be able to meet the continued listing requirements for Nasdaq or another nationally recognized stock exchange, which could limit investors’ ability to make transactions in our securities and subject us to additional trading restrictions.
In order to remain listed on Nasdaq, we will be required to meet the continued listing requirements of Nasdaq or any other U.S. or nationally recognized stock exchange to which we may apply and be approved for listing. We may be unable to satisfy these continued listing requirements, and there is no guarantee that our Common Stock will remain listed on Nasdaq or any other U.S. or nationally recognized stock exchange. If, after listing, our Common Stock is delisted from Nasdaq or any other U.S. or nationally recognized stock exchange, we could face significant material adverse consequences, including:
| · | a limited availability of market quotations for our Common Stock; |
| · | reduced liquidity with respect to the market for our Common Stock; |
| · | a determination that our Common Stock is a “penny stock,” which will require brokers trading in our Common Stock to adhere to different rules, possibly resulting in a reduced level of trading activity in the secondary trading market for our Common Stock; |
| · | a limited amount of news and analyst coverage; and |
| · | decreased ability to issue additional shares of our Common Stock or obtain additional financing in the future. |
A large number of shares may be issued and subsequently sold upon the exercise of existing options and warrants.
As of November 18, 2021, there were 42,667 shares of Common Stock issuable under outstanding options and 10,773,829 shares issuable upon exercise of outstanding warrants at various exercise prices. To the extent that holders of existing options or warrants sell the shares of Common Stock issued upon the exercise of warrants, the market price of our Common Stock may decrease due to the additional selling pressure in the market. The risk of dilution from issuances of shares of Common Stock underlying existing options and warrants may cause shareholders to sell their Common Stock, which could further decline in the market price.
| 25 |
You may experience future dilution as a result of future equity offerings.
In order to raise additional capital, we may in the future offer additional Common Stock or other securities convertible into or exchangeable for our Common Stock at prices that may not be the same as the price per share in this offering. We may sell shares or other securities in any other offering at a price per share that is less than the price per share paid by investors in this offering, and investors purchasing shares or other securities in the future could have rights superior to existing shareholders. The price per share at which we sell additional shares of our Common Stock, or securities convertible or exchangeable into Common Stock, in future transactions may be higher or lower than the price per share paid by investors in this offering.
As a newly listed company on Nasdaq, we will incur materially increased costs and be subject to additional regulations and requirements.
As a newly exchange-listed public company on Nasdaq, we will incur material additional legal, accounting and other expenses, including payment of annual exchange fees, to satisfy the continued listing standards for Nasdaq. If our Common Stock is listed on Nasdaq, we must meet certain financial and liquidity criteria to maintain our listing. If we fail to meet any of Nasdaq’s listing standards, our Common Stock may be delisted. In addition, our Board may determine that the cost of maintaining our listing on a national securities exchange outweighs the benefits of such listing. A delisting of our Common Stock from Nasdaq may materially impair our stockholders’ ability to buy and sell our Common Stock and could have an adverse effect on the market price of, and the efficiency of the trading market for, our Common Stock. The delisting of our Common Stock could significantly impair our ability to raise capital and the value of your investment.
We may experience volatility in our stock price, which may adversely affect the trading price of our Common Stock.
We have experienced significant volatility from time to time in the market price of our shares of Common Stock. Over the past twelve months, shares of our Common Stock were quoted and traded at a high of $55.22 per share and a low of $2.25 per share. In the future, the market price of our Common Stock may continue to be volatile.
Risks Related to our Reverse Stock Split
We cannot assure you that we will be able to continue to comply with the minimum bid price requirement of Nasdaq.
There can be no assurance that the market price of our Common Stock will remain at the level required for continuing compliance with Nasdaq’s minimum bid requirement. It is not uncommon for the market price of a company’s Common Stock to decline in the period following a reverse stock split. In any event, other factors unrelated to the number of shares of our Common Stock outstanding, such as negative financial or operational results, could adversely affect the market price of our Common Stock and jeopardize our ability to maintain compliance with Nasdaq’s minimum bid price requirement.
There can be no assurance that we will be able to comply with the continued listing standards of Nasdaq, a failure of which could result in a de-listing of our Common Stock.
Nasdaq requires that the trading price of its listed stocks remain above one dollar in order for the stock to remain listed. If a listed stock trades below one dollar for more than 30 consecutive trading days, then it is subject to delisting from Nasdaq. In addition, to maintain a listing on Nasdaq, we must satisfy minimum financial and other continued listing requirements and standards, including those regarding director independence and independent committee requirements, minimum stockholders’ equity and certain corporate governance requirements. If we are unable to satisfy these requirements or standards, we could be subject to delisting. This would have a negative effect on the price of our Common Stock and would impair your ability to sell or purchase our Common Stock when you wish to do so. In the event of a delisting, we can provide no assurance that any action we may take to restore our compliance with the listing requirements would allow our Common Stock to become listed again, stabilize the market price or improve the liquidity of our Common Stock, prevent our Common Stock from dropping below the minimum bid price requirement, or prevent future non-compliance with the listing requirements.
| 26 |
Risks Related to our Tax Matters
Our ability to use our net operating loss carryforwards and certain other tax attributes may be limited.
As of April 30, 2021, we had federal net operating loss carryforwards of approximately $48 million, which began to expire in varying amounts beginning in 2020. Under Sections 382 and 383 of the United States Internal Revenue Code of 1986, as amended, or the Code, and corresponding provisions of state law, if a corporation undergoes an “ownership change” (generally defined as a greater than 50-percentage-point cumulative change (by value) in the equity ownership of certain stockholders over a rolling three-year period), the corporation’s ability to use its pre-change net operating loss carryforwards and other pre-change tax attributes to offset its post-change taxable income or taxes may be limited. We may have experienced ownership changes in the past and could experience one or more ownership changes in the future, including in connection with this offering, some of which are outside our control. Our net operating loss carryforwards may also be subject to limitation under state laws. Further, our ability to utilize net operating loss carryforwards of companies that we may acquire in the future may also be subject to limitations. There is also a risk that due to tax law changes, such as suspensions on the use of net operating loss carryforwards, or other unforeseen reasons, our ability to use our pre-change net operating loss carryforwards and other pre-change tax attributes to offset post-change taxable income or taxes may be subject to limitation or expire.
Changes in U.S. tax law could adversely affect our business and financial condition.
The laws, rules and regulations dealing with U.S. federal, state, and local income taxation are constantly under review by persons involved in the legislative process and by the Internal Revenue Service and the U.S. Treasury Department. Changes to tax laws (which changes may have retroactive application) could adversely affect us or holders of our Common Stock. In recent years, many changes have been made to applicable tax laws and changes are likely to continue to occur in the future.
For example, the Tax Cuts and Jobs Act, or the TCJA, was enacted in 2017 and made significant changes to corporate taxation, including the reduction of the corporate tax rate from a top marginal rate of 35% to a flat rate of 21%, the limitation of the tax deduction for net interest expense to 30% of adjusted taxable income (except for certain small businesses), the limitation of the deduction for net operating losses from taxable years beginning after December 31, 2017 to 80% of current year taxable income and the elimination of net operating loss carrybacks generated in taxable years ending after December 31, 2017 (though any such net operating losses may be carried forward indefinitely), and the modification or repeal of many business deductions and credits. In addition, on March 27, 2020, then President Trump signed into law the “Coronavirus Aid, Relief, and Economic Security Act” or the CARES Act, which, among other things, suspends the 80% limitation on the deduction for net operating losses arising in taxable years beginning before January 1, 2021, permits a five-year carryback of net operating losses arising in taxable years beginning after December 31, 2017 and before January 1, 2021, and generally modifies the limitation on the deduction for net interest expense to 50% of adjusted taxable income for taxable years beginning in 2019 and 2020.
It cannot be predicted whether, when, in what form, or with what effective dates, new tax laws may be enacted, or regulations and rulings may be enacted, promulgated or issued under existing or new tax laws, which could result in an increase in our or our shareholders’ tax liability or require changes in the manner in which we operate in order to minimize or mitigate any adverse effects of changes in tax law or in the interpretation thereof.
We will not receive any proceeds from the sale of the shares of Common Stock by the Selling Stockholders. However, we will receive proceeds from the exercise of the Warrants by the Selling Stockholders to the extent they are exercised for cash. We estimate that the maximum proceeds that we may receive from the exercise of the Warrants, assuming all the Series A Warrants are exercised at their exercise price of $5.00 and all the Placement Agent Warrants are exercised at their exercise price of $6.25 will be $41,562,500. We do not know, however, whether any of the Warrants will be exercised or, if any of the Warrants are exercised, when they will be exercised. It is possible that the Warrants will expire and never be exercised. Further, in the event the registration statement in which this prospectus forms a part does not stay effective, the Warrants may be exercised on a cashless basis. In these circumstances, even if the Warrants are exercised, we may not receive any proceeds, or the proceeds that we do receive may be less than what we might expect.
| 27 |
We intend to use the aggregate net proceeds from the exercise of the Warrants by the Selling Stockholders to the extent they are exercised for cash (in addition to the existing cash of the Company) (i) to fully fund the Phase 2b clinical trial in LAPC, if and when the clinical hold on the IND is lifted; (ii) to continue development of the Company’s Cancer Program; (iii) to continue development of the Company’s Diabetes Program; (iv) to continue development of the Company’s Malignant Ascites Program; (v) and to fund general corporate purposes.
The amounts and timing of our use of the net proceeds from this offering will depend on a number of factors, such as the timing and progress of our efforts to lift the FDA’s clinical hold on our IND for a clinical trial for LAPC, our ability to conduct the clinical trial for LAPC if and when the FDA’s clinical hold is lifted, our R&D efforts, the timing and progress of any collaborative or strategic partnering efforts, technological advances and the competitive environment for our planned products. As of the date of this prospectus, we cannot specify with certainty the amount or timing of the proceeds the Company will receive from the exercise of the Warrants. Accordingly, our management will have broad discretion in the timing and application of these proceeds. Pending application of the net proceeds as described above, we intend to temporarily invest the proceeds in short-term, interest-bearing instruments.
The Selling Stockholders will pay any expenses incurred by the Selling Stockholders for brokerage, accounting, tax or legal services or any other expenses incurred by the Selling Stockholders in disposing of their shares of Common Stock. We will bear all other costs, fees and expenses incurred in effecting the registration of the shares covered by this prospectus, including, without limitation, all registration fees and fees and expenses of our counsel and our accountants.
We have never declared or paid any cash dividends on our Common Stock. We do not anticipate paying any cash dividends to stockholders in the foreseeable future. In addition, any future determination to pay cash dividends will be at the discretion of our Board and will be dependent upon our financial condition, results of operations, capital requirements, and such other factors as our Board deems relevant.
On August 19, 2021, the Company entered into a securities purchase agreement (“Purchase Agreement”) with certain institutional investors (“Purchasers”). Under the Purchase Agreement, and pursuant to an exemption from the registration requirements of Section 5 of the Securities Act contained in Section 4(a)(2) thereof and/or Regulation D thereunder, the Company sold Series A Warrants to purchase 7,000,000 shares of Common Stock. The Selling Stockholders were the Purchasers of the Series A Warrants under the Purchase Agreement. Additionally, pursuant to its engagement letter with Wainwright, the Company also issued 1,050,000 Placement Agent Warrants to Wainwright or its designees.
The quantity, issue date, exercise price and expiration date of the Series A Warrants and Placement Agent Warrants, respectively, are listed in the table below:
| Quantity | Issuance Date | Exercise Price | Expiration Date | ||||||
| 7,000,0000 | August 23, 2021 | $ | 5.00 | August 23, 2026 | |||||
| 1,050,000 | August 23, 2021 | $ | 6.25 | August 23, 2026 | |||||
| 28 |
Each Series A Warrant is exercisable for one share of Common Stock beginning on the date of issuance thereof and ending on the five-year anniversary of such date. The Series A Warrants have an exercise price of $5.00 per share. The exercise price and number of shares of Common Stock issuable upon exercise of the Series A Warrants are subject to adjustment in the event of any stock dividend, split, recapitalization, reorganization or similar transaction, as described in the Series A Warrants. Subject to limited exceptions, a holder of a Series A Warrant will not have the right to exercise any portion of its Series A Warrant if the holder, together with its affiliates, would beneficially own in excess of 4.99%, or at the election of the holder 9.99%, of the number of shares of Common Stock outstanding immediately after giving effect to such exercise (“Beneficial Ownership Limitation”); provided that upon 61 days’ prior notice to the Company, the holder may elect to increase or decrease the Beneficial Ownership Limitation, although in no event may the Beneficial Ownership Limitation exceed 9.99%.
In the event of any fundamental transaction, as described in the Series A Warrants and generally including any merger with or into another entity, sale of all or substantially all of our assets, tender offer or exchange offer, or reclassification of our shares of Common Stock, then upon any subsequent exercise of a Series A Warrant, the holder will have the right to receive as alternative consideration, for each share of Common Stock that would have been issuable upon such exercise immediately prior to the occurrence of such fundamental transaction, the number of shares of Common Stock of the successor or acquiring corporation of our company, if it is the surviving corporation, and any additional consideration receivable upon or as a result of such transaction by a holder of the number of shares of Common Stock for which the Series A Warrant is exercisable immediately prior to such event. Notwithstanding the foregoing, in the event of a fundamental transaction, the holders of the Series A Warrants have the right to require us or a successor entity to redeem the Series A Warrants for cash in the amount of the Black Scholes Value (as defined in each Series A Warrant) of the unexercised portion of the Series A Warrants concurrently with or within 30 days following the consummation of a fundamental transaction. However, in the event of a fundamental transaction which is not in our control, including a fundamental transaction not approved by our board of directors, the holders of the Series A Warrants will only be entitled to receive from us or our successor entity, as of the date of consummation of such fundamental transaction the same type or form of consideration (and in the same proportion), at the Black Scholes Value of the unexercised portion of the Series A Warrant, that is being offered and paid to the holders of our Common Stock in connection with the fundamental transaction, whether that consideration is in the form of cash, stock or any combination of cash and stock, or whether the holders of our Common Stock are given the choice to receive alternative forms of consideration in connection with the fundamental transaction.
Each Placement Agent Warrant is exercisable for one share of Common Stock beginning on the date of commencement of sales in the Registered Direct Offering and ending on the five-year anniversary of such date. The Placement Agent Warrants have an exercise price of $6.25 per share (which represents 125% of the offering price per Share in the Registered Direct Offering). The Placement Agent Warrants will terminate five years after the date of commencement of sales in the Offerings. The exercise price and number of shares of Common Stock issuable upon exercise of the Placement Agent Warrants are subject to adjustment in the event of any stock dividend, split, recapitalization, reorganization or similar transaction, as described in the Placement Agent Warrants. Subject to limited exceptions, a holder of a Placement Agent Warrant will not have the right to exercise any portion of its Placement Agent Warrant if the holder, together with its affiliates, would beneficially own in excess of 4.99%, or at the election of the holder 9.99%, of the number of shares of Common Stock outstanding immediately after giving effect to such exercise (the “Beneficial Ownership Limitation”); provided that upon 61 days’ prior notice to the Company, the holder may elect to increase or decrease the Beneficial Ownership Limitation, although in no event may the Beneficial Ownership Limitation exceed 9.99%.
In the event of any fundamental transaction, as described in the Placement Agent Warrants and generally including any merger with or into another entity, sale of all or substantially all of our assets, tender offer or exchange offer, or reclassification of our shares of Common Stock, then upon any subsequent exercise of a Placement Agent Warrant, the holder will have the right to receive as alternative consideration, for each share of Common Stock that would have been issuable upon such exercise immediately prior to the occurrence of such fundamental transaction, the number of shares of Common Stock of the successor or acquiring corporation of our company, if it is the surviving corporation, and any additional consideration receivable upon or as a result of such transaction by a holder of the number of shares of Common Stock for which the Placement Agent Warrant is exercisable immediately prior to such event. Notwithstanding the foregoing, in the event of a fundamental transaction, the holders of the Placement Agent Warrants have the right to require us or a successor entity to redeem the Placement Agent Warrants for cash in the amount of the Black Scholes Value (as defined in each Placement Agent Warrant) of the unexercised portion of the Placement Agent Warrants concurrently with or within 30 days following the consummation of a fundamental transaction. However, in the event of a fundamental transaction which is not in our control, including a fundamental transaction not approved by our board of directors, the holders of the Placement Agent Warrants will only be entitled to receive from us or our successor entity, as of the date of consummation of such fundamental transaction the same type or form of consideration (and in the same proportion), at the Black Scholes Value of the unexercised portion of the Placement Agent Warrant, that is being offered and paid to the holders of our Common Stock in connection with the fundamental transaction, whether that consideration is in the form of cash, stock or any combination of cash and stock, or whether the holders of our Common Stock are given the choice to receive alternative forms of consideration in connection with the fundamental transaction.
| 29 |
The Warrants will not be registered nor listed on any exchange. If at the time of exercise of the Warrants there is no effective registration statement registering, or the prospectus contained therein is not available for the issuance of the Common Stock underlying the Warrants to the applicable Selling Stockholder, then such Warrant may also be exercised, in whole or in part, at such time by means of a “cashless exercise” in which the Selling Stockholder will be entitled to receive a number of shares of Common Stock as determined by the terms of the Warrant.
There is no established trading market for the Warrants. We do not intend to list the Warrants on any securities exchange or nationally recognized trading system.
The foregoing description of the Warrants is qualified in its entirety by reference to the Form of Series A Common Warrant Purchase Warrant and the Form of Placement Agent Warrant, which are included as Exhibits 4.2 and 4.3, respectively, to this registration statement and are incorporated by reference to the Company’s Form 8-K, filed with the SEC on August 23, 2021.
We have prepared this prospectus to allow the Selling Stockholders we have identified herein, including their transferees, pledgees, donees and successors in interest, to offer for resale up to 8,050,000 shares of our Common Stock (assuming exercise of all Warrants).
The Common Stock being offered by the Selling Stockholders is that issuable to the Selling Stockholders upon exercise of the Warrants. For additional information regarding the issuances of those shares of Common Stock and Warrants, see “Private Placement of Warrants” above. We are registering the Shares of Common Stock in order to permit the Selling Stockholders to offer the Common Stock for resale from time to time.
The registration of the sale of shares of Common Stock held by the Selling Stockholders does not mean that they will sell or otherwise dispose of all or any of those securities. The Selling Stockholders may sell or otherwise dispose of all, a portion or none of such shares from time to time. See “Plan of Distribution.” We do not know the number of shares, if any, that will be offered for sale or other disposition by any of the Selling Stockholders under this prospectus. Furthermore, the Selling Stockholders may have sold, transferred or disposed of the shares of Common Stock covered hereby in transactions exempt from the registration requirements of the Securities Act since the date on which we filed this prospectus. As a result, we cannot estimate the number of shares of Common Stock each of the Selling Stockholders will beneficially own after termination of sales under this prospectus. In addition, each of the Selling Stockholders may have sold, transferred or otherwise disposed of all or a portion of its shares of Common Stock since the date on which it provided information for the table below.
This prospectus generally covers the resale of the maximum number of shares of Common Stock issuable upon exercise of the Warrants issued pursuant to the Purchase Agreement, determined as if the outstanding Warrants were exercised in full as of the trading day immediately preceding the date this registration statement was initially filed with the SEC, without regard to any limitations on the exercise of the Warrants.
| 30 |
Except as otherwise described in this prospectus, none of the Selling Stockholders has, or within the past three years has had, any position, office or other material relationship with us or any of our affiliates. Except for Wainwright or its designees, none of the Selling Stockholders is a broker-dealer or an affiliate of a broker-dealer.
Wainwright served as our exclusive placement agent in connection with the Offerings. Pursuant to that engagement letter, dated as of April 26, 2021, between the Company and Wainwright, we agreed to pay Wainwright a placement agent fee of 7.5% of the aggregate gross proceeds raised in the Offerings. In addition, the company agreed to issue to Wainwright or its designees upon closing of the Offerings, the Placement Agent Warrants. Each of Michael Vasinkevich, Noam Rubinstein, Craig Schwabe and Charles Worthman are associated persons of Wainwright and received Placement Agent Warrants as Wainwright’s designee.
The table below sets forth certain information with respect to each Selling Stockholder, including: (i) the name of each Selling Stockholder; (ii) the number of shares of our Common Stock beneficially owned by each Selling Stockholder before this offering; (iii) the maximum number of shares being offered by each Selling Stockholder pursuant to this prospectus; and (iv) each Selling Stockholder’s beneficial ownership after completion of this offering, assuming that all of the shares covered hereby (but no other shares, if any, held by the Selling Stockholders) are sold.
The table is based on information supplied to us by the Selling Stockholders or in Schedules 13G or 13D and other public documents filed with the SEC, with beneficial ownership and percentage ownership determined in accordance with the rules and regulations of the SEC, and includes information with respect to voting or investment power with respect to shares of stock. This information does not necessarily indicate beneficial ownership for any other purpose.
The percentage of each Selling Stockholder’s ownership before and after this offering is based on 20,715,804 shares of Common Stock outstanding as of November 18, 2021 are as follows:
| 31 |
| Name and Address of Selling Stockholders | No. of Shares of Common Stock Beneficially Owned Prior to this Offering1, 2 | No. of Shares of Common Stock Offered by Selling Stockholders1 | No. and Percentage of Outstanding Shares of Common Stock Beneficially Owned Subsequent to this Offering21 |
| Alpha Capital Anstalt3 | 545,716 | 250,00019 | 295,716, 1.41% |
| Alto Opportunity Master Fund, SPC - Segregated Master Portfolio B4 | 1,033,718 | 800,00019 | 1,033,718, 4.99% |
| Anson East Master Fund LP5 | 125,000 | 125,00019 | 0 |
| Anson Investments Master Fund LP5 | 492,647 | 375,00019 | 117,64722 |
| Armistice Capital Master Fund Ltd.6 | 1,500,000 | 1,500,00019 | 0 |
| Bigger Capital Fund, LP7 | 125,000 | 125,00019 | 0 |
| Boothbay Absolute Return Strategies, LP8 | 132,480 | 132,48019 | 0 |
| Boothbay Diversified Alpha Master Fund LP8 | 67,520 | 67,52019 | 0 |
| Charles Worthman9 | 13,147 | 10,50020 | 2,64722 |
| Craig Schwabe9 | 44,372 | 35,43820 | 8,93422 |
| District 2 Capital Fund LP7 | 125,000 | 125,00019 | 0 |
| Hudson Bay Master Fund Ltd.10 | 250,000 | 250,00019 | 0 |
| Intracoastal Capital LLC11 | 623,647 | 500,00019 | 123,64722 |
| Ionic Ventures, LLC12 | 1,033,718 | 400,00019 | 800,000, 3.79% |
| Iroquois Capital Investment Group LLC13 | 75,000 | 75,00019 | 0 |
| Iroquois Master Fund Ltd13 | 175,000 | 175,00019 | 0 |
| KBB Asset Management14 | 431,353 | 250,00019 | 181,35322 |
| Kingsbrook Opportunities Master Fund LP15 | 50,000 | 50,00019 | 0 |
| Michael Vasinkevich9 | 843,055 | 673,31220 | 169,74322 |
| Noam Rubinstein9 | 414,132 | 330,75020 | 83,38222 |
| S.H.N Financial Investments LTD16 | 400,000 | 300,00019 | 100,00022 |
| Sabby Volatility Warrant Master Fund, Ltd.17 | 1,033,718 | 1,250,00019 | 586,504, 2.67% |
| The Eleven Fund LLC18 | 750,000 | 250,00019 | 500,000, 2.38% |
1 Includes the Common Stock issued under the applicable Warrant agreement. The Warrant agreements include a provision that prevents the holder from exercising any portion of such Warrant agreement that would result in the holder owning more than 4.99% (9.99% in the case of Armistice Capital Master Fund Ltd.) of the Company’s Common Stock.
2 We have assumed that the Selling Stockholders will not acquire beneficial ownership of any additional Common Stock issued by us during the Offering.
| 32 |
3 Konrad Ackermann has sole voting and dispositive power over the securities held for the account of this selling stockholder. The selling stockholder’s address c/o LH Financial Services Corp., 510 Madison Ave., Suite 1400, New York, NY 10022.
4 Ayrton Capital LLC, the investment manager to Alto Opportunity Master Fund, SPC - Segregated Master Portfolio B, has discretionary authority to vote and dispose of the shares held by Alto Opportunity Master Fund, SPC - Segregated Master Portfolio B and may be deemed to be the beneficial owner of these shares. Waqas Khatri, in his capacity as Managing Member of Ayrton Capital LLC, may also be deemed to have investment discretion and voting power over the shares held by Alto Opportunity Master Fund, SPC - Segregated Master Portfolio B. Ayrton Capital LLC and Mr. Khatri each disclaim any beneficial ownership of these shares. The address of Ayrton Capital LLC is 55 Post Rd West, 2nd Floor, Westport, CT 06880.
5 Anson Advisors Inc and Anson Funds Management LP, the Co-Investment Advisers of Anson East Master Fund LP (“Anson East”) and Anson Investments Master Fund LP (“Anson Investments”), hold voting and dispositive power over the Common Shares held by Anson East and Anson Investments. Bruce Winson is the managing member of Anson Management GP LLC, which is the general partner of Anson Funds Management LP. Moez Kassam and Amin Nathoo are directors of Anson Advisors Inc. Mr. Winson, Mr. Kassam and Mr. Nathoo each disclaim beneficial ownership of these Common Shares except to the extent of their pecuniary interest therein. The principal business address of Anson East and Anson Investments is Walkers Corporate Limited, Cayman Corporate Centre, 27 Hospital Road, George Town, Grand Cayman KY1-9008, Cayman Islands.
6 Armistice Capital LLC (“Armistice Capital”) is the investment manager of Armistice Capital Master Fund Ltd. (“Master Fund”), which holds voting and dispositive power over the Common Shares held by this selling stockholder. The Master Fund’s Series A Warrants are subject to a beneficial ownership limitation that prevents the Master Fund from exercising any portion of the warrants if such exercise would result in the Master Fund owning more than 9.99% of our outstanding common stock. Steven Boyd is the Managing Member of Armistice Capital. Armistice Capital and Mr. Boyd disclaim beneficial ownership of the securities except to the extent of their respective pecuniary interests therein. The principal business address of Armistice Capital Master Fund Ltd. is 510 Madison Avenue, 7th Floor, New York, NY 10022.
7 Bigger Capital Fund GP, LLC (“Bigger GP”) is a general partner of Bigger Capital Fund, LP (“Bigger Capital”) and District 2 Capital LP (“District 2”) is the investment manager of District 2 Capital Fund LP (“District 2 CF”). Michael Bigger is the managing member of Bigger GP and District 2 and District 2 Holdings LLC (“District 2 Holdings”), which is the managing member of District 2 GP LLC (“District 2 GP”), the general partner of District 2 CF. Therefore, Mr. Bigger, District 2, District 2 Holdings and District 2 CF may be deemed to be the beneficial owner, and have the shared power to dispose of or direct the disposition, of the shares reported as beneficially owned by District 2 CF and Mr. Bigger and Bigger GP may be deemed to be the beneficial owner, and have the shared power to dispose of or direct the disposition, of the shares reported as beneficially owned by Bigger Capital and District 2 CF. The business address for Bigger Capital and District 2 CF is 11434 Glowing Sunset LN, Las Vegas, NV 89135.
8 Boothbay Diversified Alpha Master Fund LP, a Cayman Islands limited partnership (“BBDAMF”) and Boothbay Diversified Alpha Master Fund LP, a Cayman Islands limited partnership (“BBDAMF”), are managed by Boothbay Fund Management, LLC, a Delaware limited liability company (“Boothbay”). Boothbay, in its capacity as the investment manager of BBARS and BBDAMF, has the power to vote and the power to direct the disposition of all securities held by BBARS and BBDAMF. Ari Glass is the Managing Member of Boothbay. Each of BBARS, BBDAMF, Boothbay and Mr. Glass disclaim beneficial ownership of these securities, except to the extent of any pecuniary interest therein. The mailing address is c/o Kingsbrook Partners LP, 689 Fifth Avenue, 12th Floor, New York, NY 10022
9 The selling stockholder was issued Placement Agent Warrants as a designee of Wainwright in connection with the Offerings. Each selling stockholder has sole voting and dispositive power over the securities held. The business address is c/o H.C. Wainwright & Co., LLC, 430 Park Avenue, 3rd Floor, New York, New York 10022.
10 Hudson Bay Capital Management LP, the investment manager of Hudson Bay Master Fund Ltd. has voting and investment power over these securities. Sander Gerber is the managing member of Hudson Bay Capital GP LLC, which is the general partner of Hudson Bay Capital Management LP. Each of Hudson Bay Master Fund Ltd. and Sander Gerber disclaims beneficial ownership over these securities. The address of Hudson Bay Master Fund Ltd. is 777 Third Avenue, 30th Floor, New York, N.Y. 10017
| 33 |
11 Mitchell P. Kopin and Daniel B. Asher, each of whom are managers of Intracoastal Capital LLC (“Intracoastal”), have shared voting control and investment discretion over the securities reported herein that are held by Intracoastal. As a result, each of Mr. Kopin and Mr. Asher may be deemed to have beneficial ownership (as determined under Section 13(d) of the Securities Exchange Act of 1934, as amended (the “Exchange Act”)) of the securities reported herein that are held by Intracoastal. Intracoastal’s business address is 245 Palm Trail, Delray Beach, FL 33483.
12 Ionic Ventures, LLC (“Ionic”) is the record and beneficial owner of the securities set forth in the table. Brendan O’Neil and Keith Coulston are the managers of Ionic and in such capacity have joint voting and dispositive power over shares held by Ionic. Mr. O’Neil and Mr. Coulston each disclaim beneficial ownership of the reported securities except to the extent of their pecuniary interest therein. Ionic Ventures, LLC is not a licensed broker dealer or an affiliate of a licensed broker dealer. The address of Ionic Ventures, LLC is 3053 Fillmore Street, Ste. 256, San Francisco, CA 94123.
13 Richard Abbe has the sole authority and responsibility for the investments made on behalf of Iroquois Capital Investment Group LLC (“ICIG”) as its managing member and shares authority and responsibility for the investments made on behalf of Iroquois Master Fund Ltd. (the “Iroquois Master Fund”) with Kimberly Page, each of whom is a director of the Iroquois Master Fund. As such, Mr. Abbe may be deemed to be the beneficial owner of all shares of common stock held by and underlying the securities reported herein held by Iroquois Master Fund and ICIG. The selling stockholder’s address is 125 Park Ave., 25th Floor, New York, NY 10017.
14 Steve Segal is the managing member of KBB Asset Management, LLC (“KBB”) and has voting and dispositive power over the shares held by KBB. The mailing address of KBB is 47 Calle Del Sur, Palm Coast, Florida 32137.
15 Kingsbrook Partners LP (“Kingsbrook Partners”) is the investment manager of Kingsbrook Opportunities Master Fund LP (“Kingsbrook Opportunities”) and consequently has voting control and investment discretion over securities held by Kingsbrook Opportunities. Kingsbrook Opportunities GP LLC (“Opportunities GP”) is the general partner of Kingsbrook Opportunities and may be considered the beneficial owner of any securities deemed to be beneficially owned by Kingsbrook Opportunities. KB GP LLC (“GP LLC”) is the general partner of Kingsbrook Partners and may be considered the beneficial owner of any securities deemed to be beneficially owned by Kingsbrook Partners. Ari J. Storch, Adam J. Chill and Scott M. Wallace are the sole managing members of Opportunities GP and GP LLC and as a result may be considered beneficial owners of any securities deemed beneficially owned by Opportunities GP and GP LLC. Each of Kingsbrook Partners, Opportunities GP, GP LLC and Messrs. Storch, Chill and Wallace disclaim beneficial ownership of these securities. The business address for Kingsbrook Opportunities is 689 Fifth Avenue, 12th Floor, New York, NY 10022.
16 Nir Shamir and Hadar Shamir have the voting and investment control over the securities held by S.H.N Financial Investments Ltd. The address of S.H.N Financial Investments Ltd. is 3 Arik Einstein St., Herzheliya Israel.
17 Sabby Management, LLC is the investment manager of Sabby Volatility Warrant Master Fund, Ltd. (“Sabby”) and shares voting and investment power with respect to these shares in this capacity. As manager of Sabby Management, LLC, Hal Mintz also shares voting and investment power on behalf of Sabby. Each of Sabby Management, LLC and Mr. Mintz disclaims beneficial ownership over the securities listed except to the extent of their pecuniary interest therein. The principal business address of Sabby Volatility Warrant Master Fund, Ltd. and Sabby Management, LLC is 10 Mountainview Road, Suite 205, Upper Saddle River, New Jersey 07458.
18 Hart Wasko is the natural person exercising control over the securities held by the Eleven Fund LLC. The business address for the Eleven Fund LLC is 463 Adams Street, Denver, CO 80206.
19 Represents Series A Warrants.
20 Represents Placement Agent Warrants
21 Assumes all Warrants are exercised and all stock registered pursuant to this registration statement has been sold. Ownership percentage calculated based on the 20,715,804 shares of Common Stock outstanding as of November 18, 2021.
22 Percentage of share ownership is less than one percent.
| 34 |
We are registering the shares of Common Stock issuable to the Selling Stockholders upon the exercise of the Warrants to permit the resale of these shares of Common Stock by the holders of the shares of Common Stock from time to time after the date of this prospectus. We will not receive any of the proceeds from the sale by the Selling Stockholders of the shares of Common Stock. However, we will receive proceeds from the exercise of the Warrants by the Selling Stockholders to the extent they are exercised for cash. We will bear all fees and expenses incident to our obligation to register the shares of Common Stock. The Selling Stockholders, which may include donees, pledgees, transferees or other successors-in-interest selling shares of Common Stock or interests in shares of Common Stock received after the date of this prospectus from a Selling Stockholder as a gift, pledge, partnership distribution or other transfer, may sell all or a portion of the shares of Common Stock beneficially owned by them and offered hereby from time to time on any stock exchange, market or trading facility on which the shares are traded or in private transactions.
A Selling Stockholder may use any one or more of the following methods when disposing of shares or interests therein:
| · | ordinary brokerage transactions and transactions in which the broker-dealer solicits purchasers; |
| · | block trades in which the broker-dealer will attempt to sell the shares as agent, but may position and resell a portion of the block as principal to facilitate the transaction; |
| · | purchases by a broker-dealer as principal and resale by the broker-dealer for its own account; |
| · | an exchange distribution in accordance with the rules of the applicable exchange; |
| · | privately negotiated transactions; |
| · | through the writing or settlement of options or other hedging transactions, whether through an options exchange or otherwise; |
| · | through agreements between broker-dealers and the Selling Stockholders to sell a specified number of such shares at a stipulated price per share; |
| · | a combination of any such methods of sale; and |
| · | any other method permitted by applicable law. |
The Selling Stockholders may, from time to time, pledge or grant a security interest in some or all of the shares of Common Stock owned by them and, if they default in the performance of their secured obligations, the pledgees or secured parties may offer and sell the shares of Common Stock, from time to time, under this prospectus, or under an amendment to this prospectus under Rule 424(b) or other applicable provision of the Securities Act amending the list of selling stockholders to include the pledgee, transferee or other successors in interest as selling stockholders under this prospectus. The Selling Stockholders also may transfer the shares of Common Stock in other circumstances, in which case the pledgees, transferees or other successors in interest will be the selling beneficial owners for purposes of this prospectus.
| 35 |
The Selling Stockholders also may resell all or a portion of the shares in open market transactions in reliance upon Rule 144 under the Securities Act, as permitted by that rule, or Section 4(a)(1) under the Securities Act, if available, rather than under this prospectus; provided that they meet the criteria and conform to the requirements of those provisions.
In connection with the sale of our Common Stock or interests therein, the Selling Stockholders may enter into hedging transactions with broker-dealers or other financial institutions, which may in turn engage in short sales of the Common Stock in the course of hedging the positions they assume. The Selling Stockholders may also sell shares of our Common Stock short and deliver these securities to close out their short positions, or loan or pledge the Common Stock to broker-dealers that in turn may sell these securities. The Selling Stockholders may also enter into options or other transactions with broker-dealers or other financial institutions or the creation of one or more derivative securities which require the delivery to each such broker-dealer or other financial institution of shares offered by this prospectus, which shares such broker-dealer or other financial institution may resell pursuant to this prospectus (as supplemented or amended to reflect such transaction).To the extent required, this prospectus may be amended or supplemented from time to time to describe a specific plan of distribution.
The aggregate proceeds to the Selling Stockholders from the sale of the Common Stock offered by them will be the purchase price of the Common Stock less discounts or commissions, if any. Each of the Selling Stockholders reserves the right to accept and, together with its agents from time to time, to reject, in whole or in part, any proposed purchase of Common Stock to be made directly or through agents. We will not receive any of the proceeds from this offering except that we will receive proceeds from the exercise of the Warrants by the Selling Stockholders to the extent they are exercised for cash.
The Selling Stockholders and any underwriters, broker-dealers or agents that participate in the sale of the Common Stock or interests therein may be “underwriters” within the meaning of Section 2(a)(11) of the Securities Act. Any discounts, commissions, concessions or profit they earn on any resale of the shares may be underwriting discounts and commissions under the Securities Act. Selling Stockholders who are “underwriters” within the meaning of Section 2(11) of the Securities Act will be subject to the prospectus delivery requirements of the Securities Act.
To the extent required, the shares of our Common Stock to be sold, the names of the selling stockholders, the respective purchase prices and public offering prices, the names of any agents, dealer or underwriter, and any applicable commissions or discounts with respect to a particular offer will be set forth in an accompanying prospectus supplement or, if appropriate, a post-effective amendment to the registration statement that includes this prospectus.
If underwriters are used in the sale, the shares of Common Stock will be acquired by the underwriters for their own account and may be resold from time to time in one or more transactions, including negotiated transactions, at a fixed public offering price or at varying prices determined at the time of sale. In connection with any such underwritten sale of shares of Common Stock, underwriters may receive compensation from the Selling Stockholders, for whom they may act as agents, in the form of discounts, concessions or commissions. If the Selling Stockholders use an underwriter or underwriters to effectuate the sale of shares of Common Stock, we and/or they will execute an underwriting agreement with those underwriters at the time of sale of those shares of Common Stock.
To the extent required by law, the names of the underwriters will be set forth in a prospectus supplement or, if appropriate, a post-effective amendment to the registration statement that includes the prospectus supplement and the accompanying prospectus used by the underwriters to sell those securities. The obligations of the underwriters to purchase those shares of Common Stock will be subject to certain conditions precedent, and unless otherwise specified in a prospectus supplement, the underwriters will be obligated to purchase all the shares of Common Stock offered by such prospectus supplement if any of such shares of Common Stock are purchased. Any public offering price and any discounts or concessions allowed or re-allowed or paid to dealers may be changed from time to time.
We have advised the Selling Stockholders that the anti-manipulation rules of Regulation M under the Exchange Act may apply to sales of shares in the market and to the activities of the Selling Stockholders and their affiliates. The Selling Stockholders may indemnify any broker-dealer that participates in transactions involving the sale of the shares against certain liabilities, including liabilities arising under the Securities Act.
| 36 |
Listing
Our Common Stock is listed on Nasdaq under the symbol “PMCB.”
Certain legal matters with respect to the validity of the Shares will be passed upon for us by Ballard Spahr LLP, Las Vegas, Nevada.
Armanino LLP, independent registered public accounting firm, has audited our consolidated financial statements included in our Annual Report on Form 10-K for the year ended April 30, 2021, as set forth in their report, dated August 9, 2021, which is incorporated by reference in the prospectus and elsewhere in this registration statement. Our consolidated financial statements are incorporated by reference in reliance on Armanino LLP’s report, given on their authority as experts in accounting and auditing.
WHERE YOU CAN FIND MORE INFORMATION
We have filed with the SEC a registration statement on Form S-3 (File No. 333-260849), of which this prospectus is a part, under the Securities Act, to register the shares of Common Stock offered by this prospectus. However, this prospectus does not contain all of the information contained in the Registration Statement. We have omitted from this prospectus some parts of the Registration Statement as permitted by the rules and regulations of the SEC. Statements in this prospectus concerning any document we have filed as an exhibit to the Registration Statement or that we otherwise filed with the SEC are not intended to be comprehensive and are qualified in their entirety by reference to these filings. In addition, we file annual, quarterly and other reports, proxy statements and other information with the SEC. Our SEC filings are available to the public over the Internet at the SEC’s website at www.sec.gov. Our Annual Report on Form 10-K, Quarterly Reports on Form 10-Q, and Current Reports on Form 8-K, including any amendments to those reports, and other information that we file with or furnish to the SEC pursuant to Section 13(a) or 15(d) of the Exchange Act can also be accessed free of charge through the Internet. These filings will be available as soon as reasonably practicable after we electronically file such material with, or furnish it to, the SEC. The SEC’s website can be found at http://www.sec.gov. In addition, we make available, for free, on or through our website copies of these reports as soon as reasonably practicable after we electronically file or furnished them to the SEC. Our website can be found at http:www.pharmacyte.com. Our website is not a part of this prospectus.
INFORMATION INCORPORATED BY REFERENCE
We have elected to incorporate certain information by reference into this prospectus. By incorporating by reference, we can disclose important information to you by referring you to other documents we have filed or will file with the SEC. The information incorporated by reference is deemed to be part of this prospectus, except for information incorporated by reference that is superseded by information contained in this prospectus. This means that you must look at all of the SEC filings that we incorporate by reference to determine if any statements in the prospectus or any document previously incorporated by reference have been modified or superseded. This prospectus incorporates by reference the documents set forth below that we have previously filed with the SEC, except in each case the information contained in such document to the extent “furnished” and not “filed”:
| · | Our Annual Report on Form 10-K for the fiscal year ended April 30, 2021, filed with the SEC on August 10, 2021. |
| · | Our Quarterly Report on Form 10-Q for the fiscal quarter ended July 31, 2021, filed with the SEC on September 14, 2021. |
| · | Our Current Report on Form 8-K, filed with the SEC on August 12, 2021. |
| · | Our Current Report on Form 8-K, filed with the SEC on August 23, 2021. |
| · | Our Current Report on Form 8-K, filed with the SEC on September 10, 2021. |
| · | The description of our Common Stock set forth in the registration statement on Form 8-A registering our Common Stock under Section 12 of the Exchange Act, which was filed with the SEC on August 2, 2021, including any amendments or reports filed for purposes of updating such description. |
| 37 |
We also incorporate by reference all documents we file in the future pursuant to Section 13(a), 13(c), 14 or 15(d) of the Exchange Act after the date of this prospectus and prior to the sale of all the securities covered by this prospectus (including all such documents filed with the SEC after the date of the initial filing of the Registration Statement that contains this prospectus and prior to effectiveness of the Registration Statement or after such effectiveness), except in each case the information contained in such document to the extent “furnished” and not “filed.”
You may obtain copies of these documents on the website maintained by the SEC at http://www.sec.gov, or from us without charge (other than exhibits to such documents, unless such exhibits are specifically incorporated by reference into such documents) by writing us at Corporate Secretary, PharmaCyte Biotech, Inc., 3960 Howard Hughes Parkway, Suite 500, Las Vegas, Nevada 89169, or visiting our website at http://www.pharmacyte.com.
Any statement contained in a document incorporated or deemed to be incorporated by reference in this prospectus shall be deemed to be modified or superseded for the purposes of this prospectus to the extent that a statement contained herein, any prospectus supplement or in any other subsequently filed document which also is or deemed to be incorporated by reference herein modifies or supersedes that statement. Any statement so modified or superseded shall not be deemed, except as so modified or superseded, to constitute a part of this prospectus.
| 38 |

PHARMACYTE BIOTECH, INC.
8,050,000 Shares
Common Stock
Offered by the Selling Stockholders
PROSPECTUS
November 19, 2021
| 39 |